Elbasvir-Grazoprevir Zepatier
Elbasvir-Grazoprevir Zepatier Glecaprevir-Pibrentasvir Mavyret
Glecaprevir-Pibrentasvir Mavyret Ledipasvir-Sofosbuvir Harvoni
Ledipasvir-Sofosbuvir Harvoni Ribavirin Copegus, Rebetol, Ribasphere
Ribavirin Copegus, Rebetol, Ribasphere Sofosbuvir Sovaldi
Sofosbuvir Sovaldi Sofosbuvir-Velpatasvir Epclusa
Sofosbuvir-Velpatasvir Epclusa Sofosbuvir-Velpatasvir-Voxilaprevir Vosevi
Sofosbuvir-Velpatasvir-Voxilaprevir VoseviBoxed Warning
WARNING: RISK OF HEPATITIS B VIRUS REACTIVATION IN PATIENTS COINFECTED WITH HCV AND HBV
Test all patients for evidence of current or prior hepatitis B virus (HBV) infection before initiating treatment with VOSEVI. HBV reactivation has been reported in HCV/HBV coinfected patients who were undergoing or had completed treatment with HCV direct-acting antivirals (DAA) and were not receiving HBV antiviral therapy. Some cases have resulted in fulminant hepatitis, hepatic failure, and death. Monitor HCV/HBV coinfected patients for hepatitis flare or HBV reactivation during HCV treatment and post-treatment follow-up. Initiate appropriate patient management for HBV infection as clinically indicated [see Warnings and Precautions (5.1)].
Recent Major Changes
1. Indications and Usage
VOSEVI is indicated for the treatment of adult patients with chronic hepatitis C virus (HCV) infection without cirrhosis or with compensated cirrhosis (Child-Pugh A) who have [see Dosage and Administration (2.2) and Clinical Studies (14)]:
- genotype 1, 2, 3, 4, 5, or 6 infection and have previously been treated with an HCV regimen containing an NS5A inhibitor.
- genotype 1a or 3 infection and have previously been treated with an HCV regimen containing sofosbuvir without an NS5A inhibitor.
- Additional benefit of VOSEVI over sofosbuvir/velpatasvir was not shown in adults with genotype 1b, 2, 4, 5, or 6 infection previously treated with sofosbuvir without an NS5A inhibitor.
2. Dosage and Administration
2.1 Testing Prior to the Initiation of Therapy
Test all patients for evidence of current or prior HBV infection by measuring hepatitis B surface antigen (HBsAg) and hepatitis B core antibody (anti-HBc) before initiating HCV treatment with VOSEVI [see Warnings and Precautions (5.1)].
2.2 Recommended Dosage
The recommended dosage of VOSEVI is one tablet, taken orally, once daily with food [see Clinical Pharmacology (12.3)]. One tablet of VOSEVI contains 400 mg of sofosbuvir, 100 mg of velpatasvir, and 100 mg of voxilaprevir. Table 1 shows the recommended treatment regimen and duration based on patient population.
| Genotype | Patients Previously Treated with an HCV Regimen Containing: | VOSEVI Duration |
|---|---|---|
| 1, 2, 3, 4, 5, or 6 | An NS5A inhibitor* | 12 weeks |
| 1a or 3 | Sofosbuvir without an NS5A inhibitor† | 12 weeks |
| ||
2.3 Renal Impairment
No dosage adjustment of VOSEVI is recommended in patients with any degree of renal impairment including patients on dialysis [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
2.4 Moderate or Severe Hepatic Impairment
VOSEVI is not recommended in patients with moderate or severe hepatic impairment (Child-Pugh B or C) due to higher exposures of voxilaprevir in these patients [see Warnings and Precautions (5.2), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
3. Dosage Forms and Strengths
4. Contraindications
VOSEVI is contraindicated with rifampin [see Drug Interactions (7.3), and Clinical Pharmacology (12.3)].
5. Warnings and Precautions
5.1 Risk of Hepatitis B Virus Reactivation in Patients Coinfected with HCV and HBV
Hepatitis B virus (HBV) reactivation has been reported in HCV/HBV coinfected patients who were undergoing or had completed treatment with HCV direct-acting antivirals, and who were not receiving HBV antiviral therapy. Some cases have resulted in fulminant hepatitis, hepatic failure, and death. Cases have been reported in patients who are HBsAg positive and also in patients with serologic evidence of resolved HBV infection (i.e., HBsAg negative and anti-HBc positive). HBV reactivation has also been reported in patients receiving certain immunosuppressant or chemotherapeutic agents; the risk of HBV reactivation associated with treatment with HCV direct-acting antivirals may be increased in these patients.
HBV reactivation is characterized as an abrupt increase in HBV replication manifesting as a rapid increase in serum HBV DNA level. In patients with resolved HBV infection reappearance of HBsAg can occur. Reactivation of HBV replication may be accompanied by hepatitis, i.e., increases in aminotransferase levels and, in severe cases, increases in bilirubin levels, liver failure, and death can occur.
Test all patients for evidence of current or prior HBV infection by measuring HBsAg and anti-HBc before initiating HCV treatment with VOSEVI. In patients with serologic evidence of HBV infection, monitor for clinical and laboratory signs of hepatitis flare or HBV reactivation during HCV treatment with VOSEVI and during post-treatment follow-up. Initiate appropriate patient management for HBV infection as clinically indicated.
5.2 Risk of Hepatic Decompensation/Failure in Patients with Evidence of Advanced Liver Disease
Postmarketing cases of hepatic decompensation/failure, including those with fatal outcomes, have been reported in patients treated with HCV NS3/4A protease inhibitor-containing regimens, including treatment with VOSEVI. Reported cases occurred in patients with baseline cirrhosis with and without moderate or severe liver impairment (Child-Pugh B or C). Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
In patients with compensated cirrhosis (Child-Pugh A) or evidence of advanced liver disease such as portal hypertension, perform hepatic laboratory testing as clinically indicated; and monitor for signs and symptoms of hepatic decompensation such as the presence of jaundice, ascites, hepatic encephalopathy, and variceal hemorrhage. Discontinue VOSEVI in patients who develop evidence of hepatic decompensation/failure.
VOSEVI is not recommended in patients with moderate to severe hepatic impairment (Child-Pugh B or C) or those with any history of prior hepatic decompensation [see Dosage and Administration (2.4), Adverse Reactions (6.2), Use in Specific Populations (8.7), and Clinical Pharmacology (12.3)].
5.3 Serious Symptomatic Bradycardia When Coadministered with Amiodarone
Postmarketing cases of symptomatic bradycardia and cases requiring pacemaker intervention have been reported when amiodarone is coadministered with a sofosbuvir-containing regimen. A fatal cardiac arrest was reported in a patient taking amiodarone who was coadministered a sofosbuvir-containing regimen (HARVONI® (ledipasvir/sofosbuvir)). Bradycardia has generally occurred within hours to days, but cases have been observed up to 2 weeks after initiating HCV treatment. Patients also taking beta blockers, or those with underlying cardiac comorbidities and/or advanced liver disease may be at increased risk for symptomatic bradycardia with coadministration of amiodarone. Bradycardia generally resolved after discontinuation of HCV treatment. The mechanism for this effect is unknown.
Coadministration of amiodarone with VOSEVI is not recommended. For patients taking amiodarone who have no other alternative viable treatment options and who will be coadministered VOSEVI:
- Counsel patients about the risk of symptomatic bradycardia.
- Cardiac monitoring in an in-patient setting for the first 48 hours of coadministration is recommended, after which outpatient or self-monitoring of the heart rate should occur on a daily basis through at least the first 2 weeks of treatment.
Patients who are taking VOSEVI who need to start amiodarone therapy due to no other alternative viable treatment options should undergo similar cardiac monitoring as outlined above.
Due to amiodarone's long half-life, patients discontinuing amiodarone just prior to starting VOSEVI should also undergo similar cardiac monitoring as outlined above.
Patients who develop signs or symptoms of bradycardia should seek medical evaluation immediately. Symptoms may include near-fainting or fainting, dizziness or lightheadedness, malaise, weakness, excessive tiredness, shortness of breath, chest pains, confusion, or memory problems [see Adverse Reactions (6.2) and Drug Interactions (7.3)].
5.4 Risk of Reduced Therapeutic Effect Due to Concomitant Use of VOSEVI with Inducers of P-gp and/or Moderate to Strong Inducers of CYP
Drugs that are inducers of P-gp and/or moderate to strong inducers of CYP2B6, CYP2C8, or CYP3A4 (e.g., St. John's wort, carbamazepine) may significantly decrease plasma concentrations of sofosbuvir, velpatasvir, and/or voxilaprevir, leading to potentially reduced therapeutic effect of VOSEVI. The use of these agents with VOSEVI is not recommended [see Drug Interactions (7.3)].
6. Adverse Reactions
The following serious adverse reactions are described below and elsewhere in labeling:
- Serious Symptomatic Bradycardia When Coadministered with Amiodarone [see Warnings and Precautions (5.3)].
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in HCV-Infected Subjects without Cirrhosis or with Compensated Cirrhosis
The adverse reactions data for VOSEVI were derived from two Phase 3 clinical trials (POLARIS-1 and POLARIS-4) that evaluated a total of 445 subjects infected with genotype 1, 2, 3, 4, 5, or 6 HCV, without cirrhosis or with compensated cirrhosis (Child-Pugh A), who received VOSEVI for 12 weeks. VOSEVI was studied in placebo- and active-controlled (sofosbuvir/velpatasvir) trials [see Clinical Studies (14.1 and 14.2)].
The proportion of subjects who permanently discontinued treatment due to adverse events was 0.2% for subjects who received VOSEVI for 12 weeks.
The most common adverse reactions (adverse events assessed as causally related by the investigator and at least 10%) were headache, fatigue, diarrhea, and nausea in subjects treated with VOSEVI for 12 weeks.
Table 2 lists adverse reactions (adverse events assessed as causally related by the investigator, all grades) observed in at least 5% of subjects receiving 12 weeks of treatment with VOSEVI in the Phase 3 clinical trials. The side-by-side tabulation is to simplify presentation; direct comparison across trials should not be made due to differing trial designs.
| POLARIS-1 | POLARIS-4 | |||
|---|---|---|---|---|
| VOSEVI 12 weeks (N=263) | Placebo 12 weeks (N=152) | VOSEVI 12 weeks (N=182) | SOF/VEL 12 weeks (N=151) | |
| Headache | 21% | 14% | 23% | 23% |
| Fatigue | 17% | 15% | 19% | 23% |
| Diarrhea | 13% | 9% | 14% | 3% |
| Nausea | 13% | 7% | 10% | 3% |
| Asthenia | 6% | 4% | 4% | 6% |
| Insomnia | 6% | 3% | 3% | 1% |
In POLARIS-1, of the subjects receiving VOSEVI who experienced adverse reactions, 99% were mild or moderate (Grade 1 or 2) in severity. In POLARIS-4, of the subjects receiving VOSEVI who experienced adverse reactions, all the reported adverse reactions were mild or moderate (Grade 1 or 2) in severity.
Less Common Adverse Reactions Reported in Clinical Trials
The following adverse reactions occurred in less than 5% of subjects without cirrhosis or with compensated cirrhosis treated with VOSEVI for 12 weeks and are included because of a potential causal relationship.
Rash: In the POLARIS-1 and POLARIS-4 trials, rash occurred in less than 1% and 2% of subjects treated with VOSEVI, respectively. Rash was reported in 1% of subjects treated with placebo in POLARIS-1 and was not reported by any subject taking sofosbuvir/velpatasvir in POLARIS-4. No serious adverse reactions of rash occurred, and all rashes were mild or moderate in severity.
Depression: In the POLARIS-1 and POLARIS-4 trials, depressed mood occurred in less than 1% and 1% of subjects treated with VOSEVI, respectively. Depressed mood was not reported by any subject taking placebo in POLARIS-1 and was reported in 1% of subjects treated with sofosbuvir/velpatasvir in POLARIS-4. No serious adverse reactions of depressed mood occurred and all events were mild or moderate in severity.
Laboratory Abnormalities
Lipase Elevations: Isolated, asymptomatic lipase elevations of greater than 3×ULN were observed in POLARIS-1 in 2% of subjects treated with VOSEVI and 3% of subjects treated with placebo, and in POLARIS-4 in 2% of subjects treated with VOSEVI and less than 1% of subjects treated with sofosbuvir/velpatasvir.
Creatine Kinase: Isolated, asymptomatic creatine kinase elevations greater than or equal to 10×ULN were reported in POLARIS-1 in 1% of subjects treated with VOSEVI and 1% of subjects treated with placebo, and in POLARIS-4 in less than 1% of subjects treated with VOSEVI and no subjects treated with sofosbuvir/velpatasvir.
Total bilirubin: Increases in total bilirubin less than or equal to 1.5×ULN were observed in subjects treated with VOSEVI due to inhibition of OATP1B1 and OATP1B3 by voxilaprevir: 4% and 6% of subjects without cirrhosis in POLARIS-1 and POLARIS-4, respectively; and 7% and 13% of subjects with compensated cirrhosis in POLARIS-1 and POLARIS-4, respectively. No subjects experienced jaundice and total bilirubin levels decreased after completing VOSEVI treatment.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of sofosbuvir-containing regimens. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hepatobiliary Disorders
Hepatic decompensation, hepatic failure with NS3/4A protease inhibitor-containing regimens [see Warnings and Precautions (5.2)].
Cardiac Disorders
Serious symptomatic bradycardia has been reported in patients taking amiodarone who initiated treatment with a sofosbuvir-containing regimen [see Warnings and Precautions (5.3) and Drug Interactions (7.3)].
7. Drug Interactions
7.1 Potential for Other Drugs to Affect VOSEVI
Sofosbuvir, velpatasvir, and voxilaprevir are substrates of drug transporters P-gp and BCRP while GS-331007 (predominant circulating metabolite of sofosbuvir) is not. Voxilaprevir is also a substrate of OATP1B1 and OATP1B3. In vitro, slow metabolic turnover of velpatasvir by CYP2B6, CYP2C8, and CYP3A4 and of voxilaprevir by CYP1A2, CYP2C8, and primarily CYP3A4 was observed.
Drugs that are inducers of P-gp and/or moderate to strong inducers of CYP2B6, CYP2C8, or CYP3A4 (e.g., St. John's wort, carbamazepine) may significantly decrease plasma concentrations of sofosbuvir, velpatasvir, and/or voxilaprevir leading to reduced therapeutic effect of VOSEVI. The use of these agents with VOSEVI is not recommended [see Warnings and Precautions (5.4) and Clinical Pharmacology (12.3)]. VOSEVI may be coadministered with P-gp, BCRP, and CYP inhibitors. The use of OATP inhibitors which may substantially increase exposure of voxilaprevir (e.g., cyclosporine) with VOSEVI is not recommended [see Clinical Pharmacology (12.3)].
7.2 Potential for VOSEVI to Affect Other Drugs
Velpatasvir and voxilaprevir are inhibitors of drug transporters P-gp, BCRP, OATP1B1, and OATP1B3. Velpatasvir is also an inhibitor of OATP2B1. Coadministration of VOSEVI with drugs that are substrates of these transporters may alter the exposure of such drugs. Coadministration of VOSEVI with BCRP substrates (e.g., methotrexate, mitoxantrone, imatinib, irinotecan, lapatinib, rosuvastatin, sulfasalazine, topotecan) is not recommended [see Clinical Pharmacology (12.3)].
7.3 Established and Potentially Significant Drug Interactions
Clearance of HCV infection with direct acting antivirals may lead to changes in hepatic function, which may impact the safe and effective use of concomitant medications. For example, altered blood glucose control resulting in serious symptomatic hypoglycemia has been reported in diabetic patients in postmarketing case reports and published epidemiological studies. Management of hypoglycemia in these cases required either discontinuation or dose modification of concomitant medications used for diabetes treatment.
Frequent monitoring of relevant laboratory parameters (e.g., International Normalized Ratio [INR] in patients taking warfarin, blood glucose levels in diabetic patients) or drug concentrations of concomitant medications such as cytochrome P450 substrates with a narrow therapeutic index (e.g., certain immunosuppressants) is recommended to ensure safe and effective use. Dose adjustments of concomitant medications may be necessary.
Table 3 provides a listing of established or potentially clinically significant drug interactions. The drug interactions described are based on studies conducted with either VOSEVI, the components of VOSEVI (sofosbuvir, velpatasvir, and/or voxilaprevir), or are predicted drug interactions that may occur with VOSEVI [see Contraindications (4), Warnings and Precautions (5.3, 5.4), and Clinical Pharmacology (12.3)].
| Concomitant Drug Class: Drug Name | Effect on Concentration† | Clinical Effect/Recommendation |
|---|---|---|
| Acid Reducing Agents: | ↓ velpatasvir | Velpatasvir solubility decreases as pH increases. Drugs that increase gastric pH are expected to decrease concentration of velpatasvir. |
| Antacids (e.g., aluminum and magnesium hydroxide) | Separate antacid and VOSEVI administration by 4 hours. | |
| H2-receptor antagonists (e.g., famotidine)‡ | H2-receptor antagonists may be administered simultaneously with or staggered from VOSEVI at a dose that does not exceed doses comparable with famotidine 40 mg twice daily. | |
| Proton-pump inhibitors (e.g., omeprazole)‡ | Omeprazole 20 mg can be administered with VOSEVI. Use with other proton pump-inhibitors has not been studied. | |
| Antiarrhythmics: | ||
| amiodarone | Effect on amiodarone, sofosbuvir, velpatasvir, and voxilaprevir concentrations unknown | Coadministration of amiodarone with VOSEVI may result in serious symptomatic bradycardia. The mechanism of this effect is unknown. Coadministration of amiodarone with VOSEVI is not recommended; if coadministration is required, cardiac monitoring is recommended [see Warnings and Precautions (5.3)]. |
| digoxin‡ | ↑ digoxin | Therapeutic concentration monitoring of digoxin is recommended when coadministered with VOSEVI. Refer to digoxin prescribing information for monitoring and dose modification recommendations for concentration increases with unclear magnitude. |
| Anticoagulants: | ||
| dabigatran etexilate‡ | ↑ dabigatran | Clinical monitoring of dabigatran is recommended when coadministered with VOSEVI. Refer to dabigatran etexilate prescribing information for dose modification recommendations in the setting of moderate renal impairment. |
| Anticonvulsants: | ||
| carbamazepine‡
phenytoin phenobarbital | ↓ sofosbuvir ↓ velpatasvir ↓ voxilaprevir | Coadministration is not recommended. |
| Antimycobacterials: | ||
| rifampin‡ | ↓ sofosbuvir ↓ velpatasvir ↑ voxilaprevir (single dose) ↓ voxilaprevir (multiple dose) | Coadministration with rifampin is contraindicated [see Contraindications (4)]. |
| rifabutin‡
rifapentine | ↓ sofosbuvir ↓ velpatasvir ↓ voxilaprevir | Coadministration is not recommended. |
| Antiretrovirals: | ||
| atazanavir‡
lopinavir | ↑ voxilaprevir | Coadministration of VOSEVI with atazanavir- or lopinavir-containing regimens is not recommended. |
| tipranavir/ritonavir | ↓ sofosbuvir ↓ velpatasvir | Coadministration is not recommended. The effect on voxilaprevir is unknown. |
| efavirenz‡ | ↓ velpatasvir ↓ voxilaprevir | Coadministration of VOSEVI with efavirenz-containing regimens is not recommended. |
| tenofovir disoproxil fumarate (tenofovir DF)‡ | ↑ tenofovir | Monitor for tenofovir-associated adverse reactions in patients receiving VOSEVI concomitantly with a regimen containing tenofovir DF. Refer to the prescribing information of the tenofovir DF-containing product for recommendations on renal monitoring. |
| Herbal Supplements: | ||
| St. John's wort | ↓ sofosbuvir ↓ velpatasvir ↓ voxilaprevir | Coadministration is not recommended. |
| HMG-CoA Reductase Inhibitors: | ||
| pravastatin‡ | ↑ pravastatin | Coadministration of VOSEVI with pravastatin has been shown to increase the concentration of pravastatin, which is associated with increased risk of myopathy, including rhabdomyolysis. Pravastatin may be administered with VOSEVI at a dose that does not exceed pravastatin 40 mg. |
| rosuvastatin‡ | ↑ rosuvastatin | Coadministration of VOSEVI with rosuvastatin may significantly increase the concentration of rosuvastatin which is associated with increased risk of myopathy, including rhabdomyolysis. Coadministration of VOSEVI with rosuvastatin is not recommended. |
| pitavastatin | ↑ pitavastatin | Coadministration with VOSEVI may increase the concentration of pitavastatin and is not recommended, due to an increased risk of myopathy, including rhabdomyolysis. |
| atorvastatin‡
fluvastatin lovastatin simvastatin | ↑ atorvastatin ↑ fluvastatin ↑ lovastatin ↑ simvastatin | Coadministration with VOSEVI may increase the concentrations of atorvastatin, fluvastatin, lovastatin, and simvastatin. Increased statin concentrations may increase the risk of myopathy, including rhabdomyolysis. Use the lowest approved statin dose. If higher doses are needed, use the lowest necessary statin dose based on a risk/benefit assessment. |
| Immunosuppressants: | ||
| cyclosporine‡ | ↑ voxilaprevir | Coadministration of voxilaprevir with cyclosporine has been shown to substantially increase the plasma concentration of voxilaprevir, the safety of which has not been established. Coadministration of VOSEVI with cyclosporine is not recommended. |
| ||
7.4 Drugs without Clinically Significant Interactions with VOSEVI
Based on drug interaction studies conducted with the components of VOSEVI (sofosbuvir, velpatasvir, and/or voxilaprevir) or VOSEVI, no clinically significant drug interactions have been observed with the following drugs [see Clinical Pharmacology (12.3)]:
- VOSEVI: cobicistat, darunavir, elvitegravir, emtricitabine, ethinyl estradiol/norgestimate, gemfibrozil, rilpivirine, ritonavir, tenofovir alafenamide, voriconazole
- Sofosbuvir/velpatasvir: dolutegravir, ketoconazole, raltegravir
- Sofosbuvir: methadone, tacrolimus
8. Use in Specific Populations
8.1 Pregnancy
Risk Summary
No adequate human data are available to establish whether or not VOSEVI poses a risk to pregnancy outcomes. In animal reproduction studies, no evidence of adverse developmental outcomes was observed with the components of VOSEVI (sofosbuvir, velpatasvir, or voxilaprevir) at exposures greater than those in humans at the recommended human dose (RHD) [see Data]. During organogenesis in the mouse, rat, and rabbit, systemic exposures (AUC) of velpatasvir were approximately 23 (mice), 4 (rats), and 0.5 (rabbits) times the exposure in humans at the RHD, while exposures of voxilaprevir were approximately 141 (rats) and 4 (rabbits) times the exposure in humans at the RHD. Exposures of the predominant circulating metabolite of sofosbuvir (GS-331007) were approximately 6 (rats) and 16 (rabbits) times the exposure in humans at the RHD. In rat pre/postnatal development studies, maternal systemic exposures (AUC) for each component of VOSEVI were approximately 7 (sofosbuvir metabolite GS-331007), 3 (velpatasvir), and 238 (voxilaprevir) times the exposure in humans at the RHD.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2–4% and 15–20%, respectively.
Data
Sofosbuvir: Sofosbuvir was administered orally to pregnant rats (up to 500 mg/kg/day) and rabbits (up to 300 mg/kg/day) on gestation days 6 to 18 and 6 to 19, respectively, and also to rats (oral doses up to 500 mg/kg/day) from gestation day 6 to lactation/post-partum day 20. No significant effects on embryo-fetal (rats and rabbits) or pre/postnatal (rats) development were observed at the highest doses tested. The systemic exposures (AUC) of the predominant circulating metabolite of sofosbuvir (GS-331007) during gestation were approximately 6 (rats) and 16 (rabbits) times the exposure in humans at the RHD.
Velpatasvir: Velpatasvir was administered orally to pregnant mice (up to 1000 mg/kg/day), rats (up to 200 mg/kg/day) and rabbits (up to 300 mg/kg/day) from gestation days 6 to 15, 6 to 17, and 7 to 20, respectively, and also to rats (oral doses up to 200 mg/kg) on gestation day 6 to lactation/post-partum day 20. No significant effects on embryo-fetal (mice, rats, and rabbits) or pre/postnatal (rats) development were observed at the highest doses tested. The systemic exposures (AUC) of velpatasvir during gestation were approximately 23 (mice), 4 (rats), and 0.5 (rabbits) times the exposure in humans at the RHD.
Voxilaprevir: Voxilaprevir was administered orally to pregnant rats (up to 100 mg/kg/day) and rabbits (up to 600 mg/kg/day) from gestation days 6 to 17, and 7 to 19, respectively, and also to rats (oral doses up to 100 mg/kg) on gestation day 6 to lactation/post-partum day 20. No significant effects on embryo-fetal (rats and rabbits) or pre/postnatal (rats) development were observed at the highest doses tested. The systemic exposures (AUC) of voxilaprevir during gestation were approximately 141 (rats) and 4 (rabbits) times the exposure in humans at the RHD.
8.2 Lactation
Risk Summary
It is not known whether the components of VOSEVI and its metabolites are present in human breast milk, affect human milk production, or have effects on the breastfed infant. When the components of VOSEVI were administered to lactating rats, GS-331007 (the predominant circulating metabolite of sofosbuvir) and velpatasvir were detected in milk, while voxilaprevir was detected in the plasma of nursing pups likely due to the presence of voxilaprevir in milk. No significant effects of any of the drugs were observed in nursing rat pups [see Data].
The developmental and health benefits of breastfeeding should be considered along with the mother's clinical need for VOSEVI and any potential adverse effects on the breastfed child from VOSEVI or from the underlying maternal condition.
Data
Sofosbuvir: No significant effects of sofosbuvir on growth or postnatal development were observed in nursing pups at the highest dose tested in rats. Maternal systemic exposure (AUC) of the predominant circulating metabolite of sofosbuvir (GS-331007) was approximately 7 times the exposure in humans at the RHD, with exposure of approximately 2% that of maternal exposure observed in nursing pups on lactation day 10. In a lactation study, sofosbuvir metabolites (primarily GS-331007) were excreted into the milk of lactating rats following administration of a single oral dose of sofosbuvir (20 mg/kg) on lactation day 2, with milk concentrations of approximately 10% that of maternal plasma concentrations observed 1 hour post-dose.
Velpatasvir: No significant effects of velpatasvir on growth or postnatal development were observed in nursing pups at the highest dose tested in rats. Maternal systemic exposure (AUC) of velpatasvir was approximately 3 times the exposure in humans at the RHD. Velpatasvir was present in the milk (approximately 173% that of maternal plasma concentrations) of lactating rats following a single oral dose of velpatasvir (30 mg/kg), and systemic exposure (AUC) in nursing pups was approximately 4% that of maternal exposure on lactation day 10.
Voxilaprevir: No significant effects of voxilaprevir on growth or postnatal development were observed in nursing pups at the highest dose tested in rats. Maternal systemic exposure (AUC) of voxilaprevir was approximately 238 times the exposure in humans at the RHD, with exposure of approximately 58% that of maternal exposure observed in nursing pups on lactation day 10.
8.4 Pediatric Use
Safety and effectiveness of VOSEVI have not been established in pediatric patients.
8.5 Geriatric Use
Clinical trials of VOSEVI included 74 subjects aged 65 and over (17% of total number of subjects in the POLARIS-1 and POLARIS-4 Phase 3 clinical trials). No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out. No dosage adjustment of VOSEVI is warranted in geriatric patients [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
No dosage adjustment of VOSEVI is recommended for patients with mild, moderate, or severe renal impairment, including ESRD requiring dialysis [see Dosage and Administration (2.3) and Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No dosage adjustment of VOSEVI is recommended for patients with mild hepatic impairment (Child-Pugh A). VOSEVI is not recommended in patients with moderate or severe hepatic impairment (Child-Pugh B or C) due to the higher exposures of voxilaprevir (up to 6-fold in non-HCV infected subjects); the safety and efficacy have not been established in HCV-infected patients with moderate or severe hepatic impairment [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)]. Postmarketing cases of hepatic decompensation/failure have been reported in these patients [see Warnings and Precautions (5.2)].
10. Overdosage
No specific antidote is available for overdose with VOSEVI. If overdose occurs the patient must be monitored for evidence of toxicity. Treatment of overdose with VOSEVI consists of general supportive measures including monitoring of vital signs as well as observation of the clinical status of the patient. Hemodialysis can efficiently remove the predominant circulating metabolite of sofosbuvir, GS-331007, with an extraction ratio of 53%. Hemodialysis is unlikely to result in significant removal of velpatasvir or voxilaprevir since velpatasvir and voxilaprevir are highly bound to plasma protein.
11. Description
VOSEVI is a fixed-dose combination tablet containing sofosbuvir, velpatasvir, and voxilaprevir for oral administration. Sofosbuvir is a nucleotide analog HCV NS5B polymerase inhibitor, velpatasvir is an NS5A inhibitor, and voxilaprevir is an NS3/4A protease inhibitor.
Each tablet contains 400 mg sofosbuvir, 100 mg velpatasvir, and 100 mg of voxilaprevir. The tablets include the following inactive ingredients: colloidal silicon dioxide, copovidone, croscarmellose sodium, lactose monohydrate, magnesium stearate, and microcrystalline cellulose. The tablets are film-coated with a coating material containing the following inactive ingredients: ferrosoferric oxide, iron oxide red, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
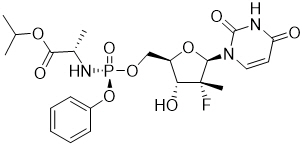
Sofosbuvir: The IUPAC name for sofosbuvir is (S)-Isopropyl 2-((S)-(((2R,3R,4R,5R)-5-(2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)-(phenoxy)phosphorylamino)propanoate. It has a molecular formula of C22H29FN3O9P and a molecular weight of 529.45. It has the following structural formula:
Sofosbuvir is a white to off-white crystalline solid with a solubility of at least 2 mg/mL across the pH range of 2–7.7 at 37 °C and is slightly soluble in water.
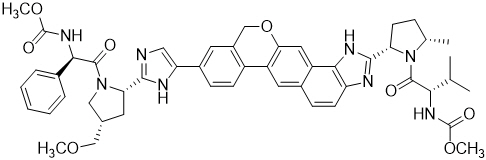
Velpatasvir: The IUPAC name for velpatasvir is Methyl {(1R)-2-[(2S,4S)-2-(5-{2-[(2S,5S)-1-{(2S)-2-[(methoxycarbonyl)amino]-3-methylbutanoyl}-5-methylpyrrolidin-2-yl]-1,11-dihydro[2]benzopyrano[4',3':6,7]naphtho[1,2-d]imidazol-9-yl}-1H-imidazol-2-yl)-4-(methoxymethyl)pyrrolidin-1-yl]-2-oxo-1-phenylethyl}carbamate. It has a molecular formula of C49H54N8O8 and a molecular weight of 883.0. It has the following structural formula:
Velpatasvir is practically insoluble (less than 0.1 mg/mL) above pH 5, slightly soluble (3.6 mg/mL) at pH 2, and soluble (greater than 36 mg/mL) at pH 1.2.
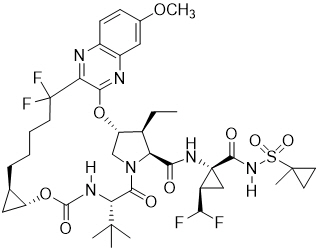
Voxilaprevir: The IUPAC name for voxilaprevir is (1aR,5S,8S,9S,10R,22aR)-5-tert-butyl-N-{(1R,2R)-2-(difluoromethyl)-1-[(1-methylcyclopropanesulfonyl) carbamoyl]cyclopropyl}-9-ethyl-18,18-difluoro-14-methoxy-3,6-dioxo-1,1a,3,4,5,6,9,10,18,19,20,21,22,22a-tetradecahydro-8H-7,10-methanocyclopropa[18,19][1,10,3,6]dioxadiazacyclononadecino[11,12-b]quinoxaline-8-carboxamide. It has a molecular formula of C40H52F4N6O9S and a molecular weight of 868.9. It has the following structural formula:
Voxilaprevir is a white to light brown solid. It is slightly hygroscopic to hygroscopic. Voxilaprevir is practically insoluble (less than 0.1 mg/mL) below pH 6.8.
12. Clinical Pharmacology
12.1 Mechanism of Action
VOSEVI is a fixed-dose combination of sofosbuvir, velpatasvir, and voxilaprevir which are DAA agents against the hepatitis C virus [see Microbiology (12.4)].
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of sofosbuvir 400 mg (recommended dosage) and 1200 mg (3 times the recommended dosage) on QTc interval was evaluated in an active-controlled (moxifloxacin 400 mg) thorough QT trial. At a dose 3 times the recommended dose, sofosbuvir does not prolong QTc to any clinically relevant extent.
The effect of velpatasvir 500 mg (5 times the recommended dosage) was evaluated in an active-controlled (moxifloxacin 400 mg) thorough QT trial. At a dose 5 times the recommended dose, velpatasvir does not prolong QTc interval to any clinically relevant extent.
The effect of voxilaprevir 900 mg (9 times the recommended dosage) was evaluated in an active-controlled (moxifloxacin 400 mg) thorough QT trial. At a dose 9 times the recommended dose, voxilaprevir does not prolong QTc interval to any clinically relevant extent.
12.3 Pharmacokinetics
The pharmacokinetic properties of the components of VOSEVI are provided in Table 4. The multiple dose pharmacokinetic parameters of sofosbuvir and its metabolite GS-331007, velpatasvir, and voxilaprevir are provided in Table 5.
| Sofosbuvir | Velpatasvir | Voxilaprevir | |
|---|---|---|---|
| CES1 = carboxylesterase 1; HINT1 = histidine triad nucleotide-binding protein 1. | |||
| |||
| Absorption | |||
| Tmax (h) | 2 | 4 | 4 |
| Effect of food (relative to fasting)* | ↑ 64% to 144% | ↑ 40% to 166% | ↑ 112% to 435% |
| Distribution | |||
| % Bound to human plasma proteins | 61–65 | >99 | >99 |
| Blood-to-plasma ratio | 0.7 | 0.5–0.7 | 0.5–0.8 |
| Metabolism | |||
| Metabolism | Cathepsin A CES1 HINT1 | CYP2B6 CYP2C8 CYP3A4 | CYP3A4 |
| Elimination | |||
| Major route of elimination | SOF: metabolism GS-331007†: glomerular filtration and active tubular secretion | Biliary excretion | Biliary excretion |
| t1/2 (h)‡ | SOF: 0.5 GS-331007†: 29 | 17 | 33 |
| % Of dose excreted in urine§ | 80¶ | 0.4 | 0 |
| % Of dose excreted in feces§ | 14 | 94 (77%# as parent) | 94 (40%# as parent) |
| Parameter Mean (%CV) | Sofosbuvir* | GS-331007† | Velpatasvir‡ | Voxilaprevir§ |
|---|---|---|---|---|
| CV = coefficient of variation; NA = not applicable. | ||||
| ||||
| Cmax
(nanogram per mL) | 678 (35.4) | 744 (28.3) | 311 (56.1) | 192 (85.8) |
| AUCtau
(nanogram∙hr per mL) | 1665 (30.1) | 12834 (29.0) | 4041 (48.6) | 2577 (73.7) |
| Ctrough
(nanogram per mL) | NA | NA | 51 (64.7) | 47 (82.0) |
Sofosbuvir and GS-331007 AUC0–24 and Cmax were similar in healthy adult subjects and subjects with HCV infection. Relative to healthy subjects (N=137), velpatasvir AUC0–24 and Cmax were 41% lower and 39% lower, respectively, in HCV-infected subjects. Relative to healthy subjects (N=63), voxilaprevir AUC0–24 and Cmax were both 260% higher in HCV-infected subjects.
Sofosbuvir and GS-331007 AUCs are near dose-proportional over the dose range of 200 mg to 1200 mg. Velpatasvir AUC increases in a greater than proportional manner from 5 to 50 mg and in a less than proportional manner from 50 to 450 mg in healthy volunteers. However, velpatasvir exhibited near dose-proportional increase in exposures 25 mg to 150 mg in HCV-infected patients. Voxilaprevir AUC increases in a greater than proportional manner over the dose range of 100 to 900 mg when administered with food.
Specific Populations
Pediatric Patients: The pharmacokinetics of VOSEVI in pediatric patients has not been established [see Use in Specific Populations (8.4)].
Geriatric Patients: Population pharmacokinetic analysis in HCV-infected subjects showed that within the age range (18 to 85 years) analyzed, age did not have a clinically relevant effect on the exposure to sofosbuvir, GS-331007, velpatasvir, or voxilaprevir [see Use in Specific Populations (8.5)].
Patients with Renal Impairment:
The pharmacokinetics of sofosbuvir were studied in HCV-negative subjects with mild (eGFR between 50 to less than 80 mL/min/1.73 m2), moderate (eGFR between 30 to less than 50 mL/min/1.73 m2), severe renal impairment (eGFR less than 30 mL/min/1.73 m2), and subjects with ESRD requiring hemodialysis following a single 400 mg dose of sofosbuvir. Relative to subjects with normal renal function (eGFR greater than 80 mL/min/1.73 m2), the sofosbuvir AUC0–inf was 61%, 107%, and 171% higher in subjects with mild, moderate, and severe renal impairment, while the GS-331007 AUC0–inf was 55%, 88%, and 451% higher, respectively. In subjects with ESRD, relative to subjects with normal renal function, sofosbuvir and GS-331007 AUC0–inf was 28% and 1280% higher when sofosbuvir was dosed 1 hour before hemodialysis compared with 60% and 2070% higher when sofosbuvir was dosed 1 hour after hemodialysis, respectively. A 4-hour hemodialysis session removed approximately 18% of administered dose of sofosbuvir [see Dosage and Administration (2.3) and Use in Specific Populations (8.6)].
The pharmacokinetics of velpatasvir were studied with a single dose of 100 mg velpatasvir in HCV-negative subjects with severe renal impairment (eGFR less than 30 mL/min by Cockcroft-Gault). No clinically relevant differences in velpatasvir pharmacokinetics were observed between healthy subjects and subjects with severe renal impairment.
The pharmacokinetics of voxilaprevir were studied with a single dose of 100 mg voxilaprevir in HCV-negative subjects with severe renal impairment (eGFR < 30 mL/min by Cockcroft-Gault). No clinically relevant differences in voxilaprevir pharmacokinetics were observed between healthy subjects and subjects with severe renal impairment.
The pharmacokinetics of sofosbuvir, GS-331007, and velpatasvir were studied in HCV-infected subjects with ESRD requiring dialysis treated with once daily sofosbuvir/velpatasvir 400/100 mg for 12 weeks. The results were consistent with those observed in HCV negative subjects with ESRD requiring dialysis. The pharmacokinetics of voxilaprevir have not been studied in subjects with ESRD. However, voxilaprevir exposure following administration of VOSEVI is not expected to be meaningfully altered in HCV-infected subjects with ESRD requiring dialysis compared to subjects with normal renal function.
Patients with Hepatic Impairment:
The pharmacokinetics of sofosbuvir were studied following 7-day dosing of 400 mg sofosbuvir in HCV-infected subjects with moderate and severe hepatic impairment (Child-Pugh B and C). Relative to subjects with normal hepatic function, the sofosbuvir AUC0–24 was 126% and 143% higher in subjects with moderate and severe hepatic impairment, respectively, while the GS-331007 AUC0–24 was 18% and 9% higher, respectively. Population pharmacokinetic analysis in HCV-infected subjects indicated that compensated cirrhosis (Child-Pugh A) had no clinically relevant effect on the exposure of sofosbuvir and GS-331007.
The pharmacokinetics of velpatasvir were studied with a single dose of 100 mg velpatasvir in HCV-negative subjects with moderate and severe hepatic impairment (Child-Pugh B and C). Velpatasvir plasma exposure (AUCinf) was similar in subjects with moderate hepatic impairment, severe hepatic impairment, and control subjects with normal hepatic function. Population pharmacokinetic analysis in HCV-infected subjects indicated that compensated cirrhosis (Child-Pugh A) had no clinically relevant effect on the exposure of velpatasvir.
The pharmacokinetics of voxilaprevir were studied with a single dose of 100 mg voxilaprevir in HCV-negative subjects with moderate and severe hepatic impairment (Child-Pugh B and C). Relative to subjects with normal hepatic function, the voxilaprevir AUCinf was 299% and 500% higher in subjects with moderate and severe hepatic impairment, respectively. Population pharmacokinetic analysis in HCV-infected subjects indicated that subjects with compensated cirrhosis (Child-Pugh A) had 73% higher exposure of voxilaprevir than those without cirrhosis [see Dosage and Administration (2.4) and Use in Specific Populations (8.7)].
Drug Interaction Studies
After oral administration of VOSEVI, sofosbuvir is rapidly absorbed and subject to extensive first-pass hepatic extraction (hydrolysis followed by sequential phosphorylation) to form the pharmacologically active triphosphate. In clinical pharmacology studies, both sofosbuvir and the primary circulating metabolite GS-331007 (dephosphorylated nucleotide metabolite) were monitored for purposes of pharmacokinetic analyses.
Sofosbuvir, velpatasvir, and voxilaprevir are substrates of drug transporters P-gp and BCRP while GS-331007 is not. Voxilaprevir, and to a lesser extent velpatasvir, are also substrates of OATP1B1 and OATP1B3. In vitro, slow metabolic turnover of velpatasvir by CYP2B6, CYP2C8, and CYP3A4 and of voxilaprevir by CYP1A2, CYP2C8, and primarily CYP3A4 was observed. Inducers of P-gp and/or moderate to strong inducers of CYP2B6, CYP2C8, or CYP3A4 (e.g., St. John's wort, carbamazepine) may significantly decrease plasma concentrations of sofosbuvir, velpatasvir, and/or voxilaprevir leading to reduced therapeutic effect of VOSEVI [see Contraindications (4), Warnings and Precautions (5.4), and Drug Interactions (7.3)]. Coadministration with drugs that inhibit P-gp and/or BCRP may increase sofosbuvir, velpatasvir, and/or voxilaprevir plasma concentrations without increasing GS-331007 plasma concentration. Coadministration with drugs that inhibit OATP may increase voxilaprevir plasma concentrations. Drugs that inhibit CYP2B6, CYP2C8, or CYP3A4 may increase plasma concentration of velpatasvir and/or voxilaprevir.
Sofosbuvir and GS-331007 are not inhibitors of drug transporters P-gp, BCRP, OATP1B1, OATP1B3, or OCT1 and GS-331007 is not an inhibitor of OAT1, OAT3, OCT2, or MATE1. Sofosbuvir and GS-331007 are not inhibitors or inducers of CYP or UGT1A1 enzymes.
Velpatasvir is an inhibitor of drug transporters P-gp, BCRP, OATP1B1, OATP1B3, and OATP2B1, and its involvement in drug interactions with these transporters is primarily limited to the process of absorption. At clinically relevant concentrations, velpatasvir is not an inhibitor of hepatic transporters OATP1A2 or OCT1, renal transporters OCT2, OAT1, OAT3 or MATE1, or CYP or UGT1A1 enzymes.
Voxilaprevir is an inhibitor of drug transporters P-gp, BCRP, OATP1B1, and OATP1B3, and its involvement in drug interactions with these transporters is primarily limited to the process of absorption. At clinically relevant concentrations, voxilaprevir is not an inhibitor of hepatic transporters OCT1, renal transporters OCT2, OAT1, OAT3, or MATE1, or CYP or UGT1A1 enzymes.
The effects of coadministered drugs on the exposure of sofosbuvir, GS-331007, velpatasvir, and voxilaprevir are shown in Table 6. The effects of sofosbuvir, velpatasvir, voxilaprevir, sofosbuvir/velpatasvir, or VOSEVI on the exposure of coadministered drugs are shown in Table 7 [see Drug Interactions (7)].
| Coadministered Drug | Sofosbuvir (SOF)/ Velpatasvir (VEL)/Voxilaprevir (VOX) | N | Geometric Mean Ratio (90% CI) of Sofosbuvir, GS-331007, Velpatasvir, and Voxilaprevir PK With/Without Coadministered Drug No Effect=1.00 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Drug | Dosage (mg) | Active Component | Dosage (mg) | ||||||||
| Component | Cmax | AUC | Cmin | ||||||||
| NA = not available/not applicable, ND = not dosed. | |||||||||||
| |||||||||||
| Atazanavir + ritonavir | 300 + 100 single dose | SOF/VEL/VOX | 400/100/100 single dose | 15 | sofosbuvir | 1.29 (1.09, 1.52) | 1.40 (1.25, 1.57) | NA | |||
| GS-331007 | 1.05 (0.99, 1.12) | 1.25 (1.16, 1.36) | NA | ||||||||
| velpatasvir | 1.29 (1.07, 1.56) | 1.93 (1.58, 2.36) | NA | ||||||||
| voxilaprevir | 4.42 (3.65, 5.35) | 4.31 (3.76, 4.93) | NA | ||||||||
| Carbamazepine | 300 twice daily | SOF | 400 single dose | 24 | sofosbuvir | 0.52 (0.43, 0.62) | 0.52 (0.46, 0.59) | NA | |||
| GS-331007 | 1.04 (0.97, 1.11) | 0.99 (0.94, 1.04) | NA | ||||||||
| Cyclosporine | 600 single dose | SOF | 400 single dose | 19 | sofosbuvir | 2.54 (1.87, 3.45) | 4.53 (3.26, 6.30) | NA | |||
| GS-331007 | 0.60 (0.53, 0.69) | 1.04 (0.90, 1.20) | NA | ||||||||
| VEL | 100 single dose | 12 | velpatasvir | 1.56 (1.22, 2.01) | 2.03 (1.51, 2.71) | NA | |||||
| VOX | 100 single dose | 25 | voxilaprevir | 19.02 (14.12, 25.62) | 9.39 (7.37, 11.96) | NA | |||||
| Darunavir + ritonavir + emtricitabine/ tenofovir DF | 800 + 100 + 200/300 once daily | SOF/VEL/VOX + VOX | 400/100/100 + 100 once daily | 29 | sofosbuvir | 0.70 (0.62, 0.78) | 0.78 (0.73, 0.83) | NA | |||
| GS-331007 | 1.06 (1.01, 1.10) | 1.15 (1.12, 1.19) | NA | ||||||||
| velpatasvir | 0.78 (0.73, 0.84) | 0.95 (0.88, 1.02) | 1.16 (1.07, 1.26) | ||||||||
| voxilaprevir | 1.72 (1.51, 1.97) | 2.43 (2.15, 2.75) | 4.00 (3.44, 4.65) | ||||||||
| Dolutegravir | 50 once daily | SOF/VEL | 400/100 once daily | 24 | sofosbuvir | 0.88 (0.80, 0.98) | 0.92 (0.85, 0.99) | NA | |||
| GS-331007 | 1.01 (0.93, 1.10) | 0.99 (0.97, 1.01) | 0.99 (0.97, 1.01) | ||||||||
| velpatasvir | 0.94 (0.86, 1.02) | 0.91 (0.84, 0.98) | 0.88 (0.82, 0.94) | ||||||||
| Efavirenz/emtricitabine/tenofovir DF† | 600/200/300 once daily | SOF/VEL | 400/100 once daily | 14 | sofosbuvir | 1.38 (1.14, 1.67) | 0.97 (0.83, 1.14) | NA | |||
| GS-331007 | 0.86 (0.80, 0.93) | 0.90 (0.85, 0.96) | 1.01 (0.95, 1.07) | ||||||||
| velpatasvir | 0.53 (0.43, 0.64) | 0.47 (0.39, 0.57) | 0.43 (0.36, 0.52) | ||||||||
| Elvitegravir/cobicistat/ emtricitabine/tenofovir alafenamide‡ | 150/150/200/ 10 once daily | SOF/VEL/VOX + VOX | 400/100/100 + 100 once daily | 29 | sofosbuvir | 1.27 (1.09, 1.48) | 1.22 (1.12, 1.32) | NA | |||
| GS-331007 | 1.28 (1.25, 1.32) | 1.43 (1.39, 1.47) | NA | ||||||||
| velpatasvir | 0.96 (0.89, 1.04) | 1.16 (1.06, 1.27) | 1.46 (1.30, 1.64) | ||||||||
| voxilaprevir | 1.92 (1.63, 2.26) | 2.71 (2.30, 3.19) | 4.50 (3.68, 5.50) | ||||||||
| Ketoconazole | 200 twice daily | VEL | 100 single dose | 12 | velpatasvir | 1.29 (1.02, 1.64) | 1.71 (1.35, 2.18) | NA | |||
| Methadone | 30 to 130 daily | SOF | 400 once daily | 14 | sofosbuvir | 0.95 (0.68, 1.33) | 1.30 (1.00, 1.69) | NA | |||
| GS-331007 | 0.73 (0.65, 0.83) | 1.04 (0.89, 1.22) | NA | ||||||||
| Omeprazole | 20 once daily 2 hours prior to VOSEVI | SOF/VEL/VOX | 400/100/100 single dose | 34 | sofosbuvir | 0.77 (0.65, 0.91) | 0.73 (0.67, 0.79) | NA | |||
| GS-331007 | 1.27 (1.20, 1.34) | 0.97 (0.94, 1.01) | NA | ||||||||
| velpatasvir | 0.43 (0.38, 0.49) | 0.46 (0.41, 0.52) | NA | ||||||||
| voxilaprevir | 0.76 (0.69, 0.85) | 0.80 (0.74, 0.87) | NA | ||||||||
| 20 once daily 4 hours after VOSEVI | SOF/VEL/ VOX | 400/100/100 single dose | 34 | sofosbuvir | 0.94 (0.83, 1.06) | 0.82 (0.77, 0.87) | NA | ||||
| GS-331007 | 1.19 (1.13, 1.26) | 0.99 (0.97, 1.01) | NA | ||||||||
| velpatasvir | 0.49 (0.43, 0.55) | 0.49 (0.43, 0.55) | NA | ||||||||
| voxilaprevir | 1.08 (0.96, 1.22) | 0.95 (0.88, 1.03) | NA | ||||||||
| Rifabutin | 300 once daily | SOF | 400 single dose | 20 | Sofosbuvir | 0.64 (0.53, 0.77) | 0.76 (0.63, 0.91) | NA | |||
| GS-331007 | 1.15 (1.03, 1.27) | 1.03 (0.95, 1.12) | NA | ||||||||
| Rifampin | 600 once daily | SOF | 400 single dose | 17 | sofosbuvir | 0.23 (0.19, 0.29) | 0.28 (0.24, 0.32) | NA | |||
| GS-331007 | 1.23 (1.14, 1.34) | 0.95 (0.88, 1.03) | NA | ||||||||
| VEL | 100 single dose | 12 | velpatasvir | 0.29 (0.23, 0.37) | 0.18 (0.15, 0.22) | NA | |||||
| VOX | 100 single dose | 24 | voxilaprevir | 0.91 (0.76, 1.10) | 0.27 (0.23, 0.31) | NA | |||||
| 600 single dose | VEL | 100 single dose | 12 | velpatasvir | 1.28 (1.05, 1.56) | 1.46 (1.17, 1.83) | NA | ||||
| VOX | 100 single dose | 24 | voxilaprevir | 11.10 (8.23, 14.98) | 7.91 (6.20, 10.09) | NA | |||||
| Tacrolimus | 5 single dose | SOF | 400 single dose | 16 | sofosbuvir | 0.97 (0.65, 1.43) | 1.13 (0.81, 1.57) | NA | |||
| GS-331007 | 0.97 (0.83, 1.14) | 1.00 (0.87, 1.13) | NA | ||||||||
| Voriconazole | 200 twice daily | VOX | 100 single dose | 24 | voxilaprevir | 1.13 (0.98, 1.31) | 1.84 (1.66, 2.03) | NA | |||
No effect on the pharmacokinetic parameters of sofosbuvir, GS-331007, velpatasvir, or voxilaprevir was observed with the combination of emtricitabine, rilpivirine, and tenofovir alafenamide; famotidine; gemfibrozil; or the combination of raltegravir, emtricitabine, and tenofovir DF.
| Coadministered Drug | Sofosbuvir (SOF)/ Velpatasvir (VEL)/Voxilaprevir (VOX) | N | Geometric Mean Ratio (90% CI) of Coadministered Drug PK With/Without Sofosbuvir, Velpatasvir, Voxilaprevir, or VOSEVI No Effect=1.00 | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Drug | Dosage (mg) | Active Component | Dosage (mg) | |||||||
| Cmax | AUC | Cmin | ||||||||
| NA = not available/not applicable. | ||||||||||
| ||||||||||
| Atorvastatin | 40 single dose | SOF/VEL | 400/100 once daily | 26 | 1.68 (1.49, 1.89) | 1.54 (1.45, 1.64) | NA | |||
| Cyclosporine | 600 single dose | SOF | 400 single dose | 19 | 1.06 (0.94, 1.18) | 0.98 (0.85, 1.14) | NA | |||
| VEL | 100 single dose | 12 | 0.92 (0.82, 1.02) | 0.88 (0.78, 1.00) | NA | |||||
| VOX | 100 single dose | 24 | 0.95 (0.88, 1.03) | 0.94 (0.84, 1.06) | NA | |||||
| Dabigatran etexilate | 75 single dose | SOF/VEL/VOX + VOX | 400/100/100 + 100 once daily | 36 | 2.87 (2.61, 3.15) | 2.61 (2.41, 2.82) | NA | |||
| Darunavir + ritonavir + emtricitabine/tenofovir DF† | darunavir 800 once daily | SOF/VEL/VOX + VOX | 400/100/100 + 100 once daily | 29 | 0.89 (0.85, 0.94) | 0.86 (0.81, 0.91) | 0.66 (0.58, 0.74) | |||
| ritonavir 100 once daily | 1.60 (1.47, 1.75) | 1.45 (1.35, 1.57) | 0.80 (0.72, 0.89) | |||||||
| emtricitabine 200 once daily | 0.88 (0.82, 0.94) | 0.99 (0.96, 1.03) | 1.20 (1.15, 1.26) | |||||||
| tenofovir DF 300 once daily | 1.48 (1.36, 1.61) | 1.39 (1.32, 1.46) | 1.47 (1.38, 1.56) | |||||||
| Digoxin | 0.25 single dose | VEL | 100 once daily | 21 | 1.88 (1.71, 2.08) | 1.34 (1.13, 1.60) | NA | |||
| Efavirenz/emtricitabine/tenofovir DF‡ | efavirenz 600 once daily | SOF/VEL | 400/100 once daily | 15 | 0.81 (0.74, 0.89) | 0.85 (0.80, 0.91) | 0.90 (0.85, 0.95) | |||
| emtricitabine 200 once daily | 1.07 (0.98, 1.18) | 1.07 (1.00, 1.14) | 1.10 (0.97, 1.25) | |||||||
| tenofovir DF 300 once daily | 1.77 (1.53, 2.04) | 1.81 (1.68, 1.94) | 2.21 (2.00, 2.43) | |||||||
| Elvitegravir/cobicistat/ emtricitabine/tenofovir alafenamide§ | elvitegravir 150 once daily | SOF/VEL/VOX + VOX | 400/100/100 + 100 once daily | 29 | 0.79 (0.75, 0.85) | 0.94 (0.88, 1.00) | 1.32 (1.17, 1.49) | |||
| cobicistat 150 once daily | 1.23 (1.18, 1.28) | 1.50 (1.44, 1.58) | 3.50 (3.01, 4.07) | |||||||
| emtricitabine 200 once daily | 0.87 (0.84, 0.91) | 0.96 (0.94, 0.99) | 1.14 (1.09, 1.20) | |||||||
| tenofovir alafenamide 10 once daily | 0.79 (0.68, 0.92) | 0.93 (0.85, 1.01) | NA | |||||||
| Emtricitabine/rilpivirine/tenofovir alafenamide¶ | emtricitabine 200 once daily | SOF/VEL/VOX + VOX | 400/100/100 + 100 once daily | 30 | 0.88 (0.83, 0.93) | 0.93 (0.90, 0.96) | 1.07 (1.01, 1.14) | |||
| rilpivirine 25 once daily | 0.79 (0.74, 0.84) | 0.80 (0.76, 0.85) | 0.82 (0.77, 0.87) | |||||||
| tenofovir alafenamide 25 once daily | 1.32 (1.17, 1.48) | 1.52 (1.43, 1.61) | NA | |||||||
| Pravastatin | 40 single dose | SOF/VEL/VOX + VOX | 400/100/100 + 100 once daily | 19 | 1.89 (1.53, 2.34) | 2.16 (1.79, 2.60) | NA | |||
| Rosuvastatin | 10 single dose | SOF/VEL/VOX + VOX | 400/100/100 + 100 once daily | 19 | 18.88 (16.23, 21.96) | 7.39 (6.68, 8.18) | NA | |||
| Raltegravir + emtricitabine/tenofovir DF | emtricitabine 200 once daily | SOF/VEL | 400/100 once daily | 30 | 1.08 (1.04, 1.12) | 1.05 (1.03, 1.07) | 1.02 (0.97, 1.08) | |||
| tenofovir DF 300 once daily | 1.46 (1.39, 1.54) | 1.40 (1.34, 1.45) | 1.70 (1.61, 1.79) | |||||||
| raltegravir 400 twice daily | 1.03 (0.74, 1.43) | 0.97 (0.73, 1.28) | 0.79 (0.42, 1.48) | |||||||
| Tacrolimus | 5 single dose | SOF | 400 once daily | 16 | 0.73 (0.59, 0.90) | 1.09 (0.84, 1.40) | NA | |||
No effect on the pharmacokinetic parameters of the following coadministered drugs was observed with VOSEVI (ethinyl estradiol/norgestimate) or its components sofosbuvir/velpatasvir (dolutegravir) or sofosbuvir (methadone).
12.4 Microbiology
Mechanism of Action
Sofosbuvir is an inhibitor of the HCV NS5B RNA-dependent RNA polymerase, which is required for viral replication. Sofosbuvir is a nucleotide prodrug that undergoes intracellular metabolism to form the pharmacologically active uridine analog triphosphate (GS-461203), which can be incorporated into HCV RNA by the NS5B polymerase and acts as a chain terminator. In a biochemical assay, GS-461203 inhibited the polymerase activity of the recombinant NS5B from HCV genotype 1b, 2a, 3a, and 4a with an IC50 value ranging from 0.36 to 3.3 µM. GS-461203 is neither an inhibitor of human DNA and RNA polymerases nor an inhibitor of mitochondrial RNA polymerase.
Velpatasvir is an inhibitor of the HCV NS5A protein, which is required for viral replication. Resistance selection in cell culture and cross-resistance studies indicate velpatasvir targets NS5A as its mode of action.
Voxilaprevir is a noncovalent, reversible inhibitor of the NS3/4A protease, which is necessary for the proteolytic cleavage of the HCV encoded polyprotein (into mature forms of the NS3, NS4A, NS4B, NS5A, and NS5B proteins) and is essential for viral replication. In a biochemical inhibition assay, voxilaprevir inhibited the proteolytic activity of recombinant NS3/4A enzymes from clinical isolates of HCV genotypes 1b and 3a with Ki values of 38 and 66 pM, respectively.
Antiviral Activity
In HCV replicon assays, sofosbuvir had median EC50 values of 15–110 nM against full-length or chimeric laboratory isolates and clinical isolates from subtypes 1a, 1b, 2a, 2b, 3a, 4a, 4d, 5a, and 6a. Velpatasvir had median EC50 values of 0.002–0.13 nM against full-length or chimeric laboratory isolates and clinical isolates from subtypes 1a, 1b, 2a, 2b, 3a, 4a, 4d, 4r, 5a, 6a, and 6e. Voxilaprevir had median EC50 values of 0.2–6.6 nM against full-length or chimeric laboratory isolates and clinical isolates from subtypes 1a, 1b, 2a, 2b, 3a, 4a, 4d, 4r, 5a, 6a, 6e, and 6n.
Evaluation of sofosbuvir in combination with velpatasvir or voxilaprevir, as well as the combination of velpatasvir and voxilaprevir, showed no antagonistic effect in reducing HCV RNA levels in replicon cells.
Resistance
In Cell Culture
HCV replicons with reduced susceptibility to sofosbuvir have been selected in cell culture for multiple genotypes including 1b, 2a, 2b, 3a, 4a, 5a, and 6a. Reduced susceptibility to sofosbuvir was associated with the nucleotide analog NS5B polymerase inhibitor resistance substitution, S282T, in all replicon genotypes examined. An M289L substitution emerged along with the S282T substitution in genotypes 2a, 5, and 6 replicons. Site-directed mutagenesis of the S282T substitution in replicons of genotype 1 to 6 conferred 2- to 18-fold reduced susceptibility to sofosbuvir.
HCV genotype 1a, 1b, 2a, 3a, 4a, 5a, and 6a replicon variants with reduced susceptibility to velpatasvir were selected in cell culture. The replicon variants developed amino acid substitutions at NS5A inhibitor resistance-associated positions 24, 28, 30, 31, 32, 58, 92, and 93. Phenotypic analysis of site-directed mutant replicons of the selected NS5A substitutions showed that single Y93H/N and the combination of L31V + Y93H/N in genotype 1a, the combination of L31V + Y93H in genotype 1b, the single substitution Y93H/S in genotype 3a, and single substitutions L31V and P32A/L/Q/R in genotype 6 conferred greater than 100-fold reduction in velpatasvir susceptibility. In the genotype 2a replicon, the single substitutions F28S and Y93H showed 91-fold and 46-fold reduced susceptibility to velpatasvir, respectively. The single substitution Y93H conferred 3-fold reduced susceptibility to velpatasvir in genotype 4a replicons. Combinations of these NS5A substitutions often showed greater reductions in susceptibility to velpatasvir than single substitutions alone.
HCV genotype 1a, 1b, 2a, 3a, 4a, 5a, and 6a replicon variants with reduced susceptibility to voxilaprevir were selected in cell culture. Amino acid substitutions were selected at NS3/4A protease inhibitor resistance-associated positions 41, 156, and 168. Site-directed mutagenesis of NS3 resistance-associated substitutions showed that substitutions conferring a greater than 100-fold reduction in voxilaprevir susceptibility were A156L/T in genotype 1a, A156T/V in genotype 1b, A156L/V in genotype 2a, A156T/V in genotype 3a, and A156L/T/V in genotype 4. Combinations of these NS3 substitutions often showed greater reductions in susceptibility to voxilaprevir than single substitutions alone.
In Clinical Trials
Of the 263 NS5A inhibitor-experienced subjects treated with VOSEVI for 12 weeks in POLARIS-1, 7 of 263 (3%) subjects (2 with genotype 1a, 4 with genotype 3a, and 1 with genotype 4d) did not achieve SVR12 and qualified for resistance analysis; 6 relapsed and 1 experienced virologic breakthrough. All the virologic failures had cirrhosis and all had a previous DAA regimen containing sofosbuvir; 3 were previously treated with ledipasvir/sofosbuvir, 2 were previously treated with sofosbuvir/velpatasvir, and 2 were previously treated with daclatasvir and sofosbuvir. Six of the 7 virologic failures had baseline NS5A inhibitor resistance-associated substitutions at position 30 or 93. All 7 virologic failures had NS5A resistance-associated substitutions at failure using a sensitivity threshold of 1% of the viral population.
Of the 2 genotype 1a virologic failure subjects, one subject with virologic breakthrough at Week 12 had virus with the NS5A resistance-associated substitution Q30T at baseline and failure and emergent NS5A resistance-associated substitutions L31M and Y93H at breakthrough; the other subject had virus with the NS5A resistance-associated substitution Y93N at baseline and relapse and emergence of low-level K24R (1.2%) in NS5A and V36A (2%) in NS3 at relapse.
Of the 4 genotype 3a virologic failure subjects, one subject had virus with emergent NS5A resistance-associated substitution E92K. Two subjects had virus with Y93H at relapse that was enriched from baseline. The remaining subject had virus with the NS5A resistance-associated substitution A30K at baseline and relapse and emergence of low-level Q41K (2%), V55A (3%), and R155M (1%) substitutions in NS3 at relapse.
The genotype 4d subject who relapsed had virus with emergent NS5A resistance-associated substitution Y93H.
No NS5B nucleotide analog inhibitor resistance-associated substitutions emerged among the virologic failure subjects from POLARIS-1.
In POLARIS-4, of the 182 DAA-experienced subjects who had not received an NS5A inhibitor treated with VOSEVI for 12 weeks, 1 subject (genotype 1a) of 182 (1%) subjects relapsed and qualified for resistance analysis. The NS5A resistance-associated substitution M28T (7.5%) emerged in this subject at relapse. No NS3/4A protease inhibitor or nucleotide analog NS5B inhibitor substitutions were observed in this subject at relapse.
Persistence of Resistance-Associated Substitutions
No data are available on the persistence of sofosbuvir, velpatasvir, or voxilaprevir resistance-associated substitutions. NS5A inhibitor resistance-associated substitutions observed with administration of other NS5A inhibitors have been found to persist for longer than 1 year in most patients. The long-term clinical impact of the emergence or persistence of virus containing sofosbuvir, velpatasvir, or voxilaprevir resistance-associated substitutions is unknown.
Effect of Baseline HCV Variants on Treatment Response
Analyses were conducted to explore the association between SVR12 rates and preexisting baseline NS3/4A protease inhibitor and NS5A inhibitor resistance-associated substitutions for subjects in POLARIS-1 and POLARIS-4. Amino acid positions considered in resistance analyses included NS3 positions 36, 41, 43, 54, 55, 56, 155, 156, and 168, and NS5A positions 24, 28, 30, 31, 58, 92, or 93. Baseline resistance-associated amino acid substitutions, which may include natural polymorphisms or prior treatment-emergent substitutions relative to subtype-specific references, were identified by next generation sequencing analysis using a sensitivity threshold of 15% of the viral population.
Overall, the presence of baseline NS3/4A protease inhibitor, NS5A inhibitor, and nucleotide analog NS5B polymerase inhibitor resistance-associated substitutions did not alter the SVR rates for DAA-experienced subjects in the POLARIS-1 and POLARIS-4 trials who received 12 weeks of VOSEVI. For subjects treated with VOSEVI for 12 weeks, SVR12 rates in subjects with or without baseline NS3 and NS5A resistance-associated substitutions in the POLARIS-1 and POLARIS-4 trials were all greater than or equal to 97%.
In POLARIS 1, which included NS5A inhibitor-experienced subjects, 79% (206/260) of subjects had baseline NS5A resistance-associated substitutions across all genotypes. The most prevalent NS5A resistance-associated substitutions were at primary resistance-associated amino acid positions 30 (97/206; 47%), 31 (58/206; 28%), and 93 (103/206; 50%). Fifty-five percent (n=113/206) of the subjects had a single NS5A resistance-associated substitution, while 2 resistance-associated substitutions were detected in 65/206 subjects (32%) and 3 or more were detected in 28/206 subjects (14%). Overall prevalence of NS3/4A protease inhibitor resistance-associated substitutions across all genotypes was 15% (37/248). The most prevalent NS3 resistance-associated substitutions were at positions 36 (5/17; 29%) and 168 (7/17; 41%) in genotype 1a and position 56 in genotype 1b (8/12; 67%). Substitutions at positions 36, 56, or 168 were detected in 1-2 subjects for each genotype 2, 3, or 4.
In POLARIS-4, which included DAA-experienced subjects who had not received an NS5A inhibitor, 32% (57/177) of subjects who received 12 weeks of VOSEVI had baseline NS5A inhibitor resistance-associated substitutions. Most of the subjects had a single NS5A resistance-associated substitution (n=40; 70%). The most prevalent NS5A resistance-associated substitution was at amino acid position 31 (n=27; 47%). Overall prevalence of baseline NS3/4A protease inhibitor resistance-associated substitutions was 12% (21/169). The most prevalent NS3 resistance-associated substitutions were at positions 55 (5/10) and 168 (3/10) in genotype 1a, position 56 in genotype 1b (3/5) and genotype 2 (3/3), and at position 168 in genotype 4 (3/3).
SVR12 was achieved in 18 of 19 (95%) subjects who had baseline nucleotide analog NS5B polymerase inhibitor resistance-associated substitutions in POLARIS-1, including 2 subjects who had virus with the S282T nucleotide analog NS5B polymerase inhibitor resistance-associated substitution in addition to NS5A resistance-associated substitutions at baseline. In POLARIS-4, a total of 14 subjects had virus with nucleotide analog NS5B polymerase inhibitor resistance-associated substitutions at baseline and all achieved SVR12.
Cross Resistance
Cross-resistance is possible between HCV NS3/4A protease inhibitors and between HCV NS5A inhibitors by class. Sofosbuvir, velpatasvir, and voxilaprevir were each active against substitutions associated with resistance to other classes of DAAs with different mechanisms of actions (e.g., voxilaprevir was fully active against virus with NS5A resistance-associated substitutions and nucleotide analog NS5B inhibitor resistance-associated substitutions).
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
Sofosbuvir: Sofosbuvir was not genotoxic in a battery of in vitro or in vivo assays, including bacterial mutagenicity, chromosome aberration using human peripheral blood lymphocytes, and in vivo mouse micronucleus assays.
Sofosbuvir was not carcinogenic in a 2-year mouse study (up to 200 mg/kg/day in males and 600 mg/kg/day in females) and in a 2-year rat study (up to 750 mg/kg/day), resulting in exposures of the predominant circulating metabolite GS-331007 of approximately 4 and 17 times (in male and female mice, respectively) and 9 times (in rats) the exposure in humans at the recommended human dose (RHD).
Velpatasvir: Velpatasvir was not genotoxic in a battery of in vitro or in vivo assays, including bacterial mutagenicity, chromosome aberration using human peripheral blood lymphocytes, and in vivo rat micronucleus assays.
Velpatasvir was not carcinogenic in a 6-month rasH2 transgenic mouse study (up to 1000 mg/kg/day) and a 2-year rat carcinogenicity study (up to 200 mg/kg/day). The exposure of VEL in the 2-year rat study was approximately 6 times the exposure in humans at the RHD.
Impairment of Fertility
Sofosbuvir: Sofosbuvir had no effects on embryo-fetal viability or on fertility when evaluated in rats. At the highest dose tested, AUC exposure to the predominant circulating metabolite GS-331007 was approximately 4 times the exposure in humans at the RHD.
14. Clinical Studies
14.1 Description of Clinical Trials
The efficacy of VOSEVI was evaluated in two Phase 3 trials in DAA-experienced subjects with genotype 1, 2, 3, 4, 5, or 6 HCV infection without cirrhosis or with compensated cirrhosis, as summarized in Table 8.
| Trial | Population | Study Arms and Comparator Groups (Number of Subjects Treated) |
|---|---|---|
| DAA: direct-acting antiviral; SOF: sofosbuvir; VEL: velpatasvir | ||
| ||
| POLARIS-1*
(NCT02607735) | Genotype 1, 2, 3, 4, 5, or 6 NS5A inhibitor-experienced†, without cirrhosis or with compensated cirrhosis | VOSEVI 12 weeks (263) Placebo 12 weeks (152) |
| POLARIS-4‡
(NCT02639247) | Genotype 1, 2, 3, or 4 DAA-experienced§ who have not received an NS5A inhibitor, without cirrhosis or with compensated cirrhosis | VOSEVI 12 weeks (182) SOF/VEL 12 weeks (151) |
Serum HCV RNA values were measured during the clinical trials using the COBAS AmpliPrep/COBAS Taqman HCV test (version 2.0) with a lower limit of quantification (LLOQ) of 15 IU/mL. Sustained virologic response (SVR12), defined as HCV RNA less than LLOQ at 12 weeks after the cessation of treatment, was the primary endpoint in both trials. Relapse is defined as HCV RNA greater than or equal to LLOQ after end-of-treatment response among subjects who completed treatment. On-treatment virologic failure is defined as breakthrough, rebound, or non-response.
14.2 Clinical Trials in HCV DAA-Experienced Subjects
NS5A Inhibitor-Experienced Adults Without Cirrhosis or With Compensated Cirrhosis (POLARIS-1)
POLARIS-1 was a randomized, double-blind, placebo-controlled trial that evaluated 12 weeks of treatment with VOSEVI compared with 12 weeks of placebo in DAA-experienced subjects with genotype 1, 2, 3, 4, 5, or 6 HCV infection without cirrhosis or with compensated cirrhosis who previously failed a regimen containing an NS5A inhibitor. Subjects with genotype 1 HCV infection were randomized 1:1 to each group. Subjects with genotype 2, 3, 4, 5, or 6 HCV infection were enrolled to the VOSEVI group. Randomization was stratified by the presence or absence of cirrhosis.
Demographics and baseline characteristics were generally balanced across treatment groups. Of the 415 treated subjects, the median age was 59 years (range: 27 to 84); 77% of the subjects were male; 81% were White; 14% were Black; 6% were Hispanic or Latino; 33% had a baseline body mass index at least 30 kg/m2; the majority of subjects had genotype 1 (72%) or genotype 3 (19%) HCV infection; 82% had a non-CC IL28B genotype (CT or TT); 74% had baseline HCV RNA levels at least 800,000 IU/mL; and 41% had compensated cirrhosis. In the POLARIS-1 trial, prior DAA regimens contained the following NS5A inhibitors: ledipasvir (51%), daclatasvir (27%), ombitasvir (11%), velpatasvir (7%), and elbasvir (3%).
Table 9 presents the SVR12 by HCV genotype for the POLARIS-1 trial. No subjects in the placebo group achieved SVR12.
| VOSEVI 12 Weeks (N=263) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Total (all GTs)* (N=263) | GT-1 | GT-2 (N=5) | GT-3 (N=78) | GT-4 (N=22) | GT-5 (N=1) | GT-6 (N=6) | ||||||
| GT-1a (N=101) | GT-1b (N=45) | Total† (N=150) | ||||||||||
| GT: genotype | ||||||||||||
| ||||||||||||
| SVR12 | 96% (253/263) | 96% (97/101) | 100% (45/45) | 97% (146/150) | 100% (5/5) | 95% (74/78) | 91% (20/22) | 100% (1/1) | 100% (6/6) | |||
| Outcome for Subjects without SVR | ||||||||||||
| On-Treatment Virologic Failure | <1% (1/263) | 1% (1/101) | 0/45 | 1% (1/150) | 0/5 | 0/78 | 0/22 | 0/1 | 0/6 | |||
| Relapse‡ | 2% (6/261) | 1% (1/100) | 0/45 | 1% (1/149) | 0/5 | 5% (4/78) | 5% (1/21) | 0/1 | 0/6 | |||
| Other§ | 1% (3/263) | 2% (2/101) | 0/45 | 1% (2/150) | 0/5 | 0/78 | 5% (1/22) | 0/1 | 0/6 | |||
DAA-Experienced Adults Without Cirrhosis or With Compensated Cirrhosis Who Had Not Received An NS5A Inhibitor (POLARIS-4)
POLARIS-4 was a randomized, open-label trial that evaluated 12 weeks of treatment with VOSEVI and 12 weeks of treatment with SOF/VEL in subjects with genotype 1, 2, 3, or 4 HCV infection without cirrhosis or with compensated cirrhosis who had previously failed a HCV DAA-containing regimen that did not include an NS5A inhibitor. Subjects whose only DAA exposure was an NS3/4A protease inhibitor were excluded. Subjects with genotype 1, 2, or 3 HCV infection were randomized 1:1 to each group. Randomization was stratified by HCV genotype and by the presence or absence of cirrhosis. Subjects with genotype 4 HCV infection were enrolled to the VOSEVI group. No subjects with genotype 5 or 6 were enrolled.
Demographics and baseline characteristics were generally balanced across treatment groups. Of the 333 treated subjects, the median age was 58 years (range: 24 to 85); 77% of the subjects were male; 87% were White, 9% were Black; 8% were Hispanic or Latino; 35% had a baseline body mass index at least 30 kg/m2; 81% had non-CC IL28B genotypes (CT or TT); 75% had baseline HCV RNA levels at least 800,000 IU/mL; and 46% had compensated cirrhosis. In the POLARIS-4 trial, prior DAA regimens contained sofosbuvir (85%) with the following: peginterferon alfa and ribavirin or ribavirin (69%), HCV NS3/4A protease inhibitor (boceprevir, simeprevir, or telaprevir; 15%), and investigational DAA (<1%). Of the 15% of subjects without prior sofosbuvir exposure, most received investigational HCV DAAs or approved HCV NS3/4A protease inhibitors, with or without peginterferon alfa and ribavirin.
Treatment with VOSEVI for 12 weeks resulted in numerically higher SVR12 rates than treatment with sofosbuvir/velpatasvir for 12 weeks in subjects with HCV genotype 1a and 3 infection. Comparable SVR12 rates were observed in subjects with HCV genotype 1b and 2 infection treated with VOSEVI for 12 weeks or with sofosbuvir/velpatasvir for 12 weeks. No comparison data are available for HCV genotypes 4, 5, and 6. Given these data, the additional benefit of VOSEVI has not been shown over sofosbuvir/velpatasvir for these genotypes and VOSEVI is only indicated for the treatment of HCV genotypes 1a or 3 infection in adults who previously received sofosbuvir without an NS5A inhibitor.
Table 10 presents the comparative virologic outcome data for HCV genotype 1, 2, and 3 subjects with prior exposure to a sofosbuvir-containing regimen.
| *Subjects with prior exposure to a SOF-containing regimen | VOSEVI 12 Weeks (N=139) | SOF/VEL 12 Weeks (N=125) |
|---|---|---|
| Overall (Genotypes 1, 2, and 3) | ||
| SVR12 | 97% (135/139) | 88% (110/125) |
| Not achieving SVR12 | ||
| On-treatment virologic failure | 0% (0/139) | 1% (1/125) |
| Relapse* | 1% (1/139) | 10% (13/124) |
| Other† | 2% (3/139) | 1% (1/125) |
| Genotype 1 | ||
| SVR12 | 96% (52/54) | 85% (34/40) |
| Not achieving SVR12 | ||
| On-treatment virologic failure | 0% (0/54) | 0% (0/40) |
| Relapse* | 2% (1/54) | 13% (5/40) |
| Other† | 2% (1/54) | 3% (1/40) |
| Genotype 1a | ||
| SVR12 | 97% (35/36) | 82% (23/28) |
| Not achieving SVR12 | ||
| On-treatment virologic failure | 0% (0/36) | 0% (0/28) |
| Relapse* | 3% (1/36) | 18% (5/28) |
| Other† | 0% (0/36) | 0% (0/28) |
| Genotype 1b | ||
| SVR12 | 94% (17/18) | 92% (11/12) |
| Not achieving SVR12 | ||
| On-treatment virologic failure | 0% (0/18) | 0% (0/12) |
| Relapse* | 0% (0/18) | 0% (0/12) |
| Other† | 6% (1/18) | 8% (1/12) |
| Genotype 2 | ||
| SVR12 | 100% (31/31) | 97% (32/33) |
| Not achieving SVR12 | ||
| On-treatment virologic failure | 0% (0/31) | 3% (1/33) |
| Relapse* | 0% (0/31) | 0% (0/32) |
| Other† | 0% (0/31) | 0% (0/33) |
| Genotype 3 | ||
| SVR12 | 96% (52/54) | 85% (44/52) |
| Not achieving SVR12 | ||
| On-treatment virologic failure | 0% (0/54) | 0% (0/52) |
| Relapse* | 0% (0/54) | 15% (8/52) |
| Other† | 4% (2/54) | 0% (0/52) |
| ||
In POLARIS-4, VOSEVI was administered for 12 weeks to 18 HCV genotype 4 subjects (with or without cirrhosis) who had prior exposure to a SOF-containing regimen without an NS5A inhibitor. All subjects achieved SVR12.
16. How Supplied/Storage and Handling
Each VOSEVI tablet contains 400 mg of sofosbuvir, 100 mg of velpatasvir, and 100 mg of voxilaprevir. The tablets are beige, capsule-shaped, film-coated, and debossed with "GSI" on one side and " " on the other side. Each bottle contains 28 tablets (NDC 61958-2401-1), polyester coil, silica gel desiccant, and is closed with a child-resistant closure.
" on the other side. Each bottle contains 28 tablets (NDC 61958-2401-1), polyester coil, silica gel desiccant, and is closed with a child-resistant closure.
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Risk of Hepatitis B Virus Reactivation in Patients Coinfected with HCV and HBV
Inform patients that HBV reactivation can occur in patients coinfected with HBV during or after treatment of HCV virus infection. Advise patients to tell their healthcare provider if they have a history of hepatitis B infection [see Warnings and Precautions (5.1)].
Risk of Hepatic Decompensation/Failure in Patients with Evidence of Advanced Liver Disease
Advise patients to seek medical evaluation immediately for symptoms of worsening liver problems such as nausea, tiredness, yellowing of the skin or white part of the eyes, bleeding or bruising more easily than normal, confusion, loss of appetite, diarrhea, dark or brown urine, dark or bloody stool, swelling of the stomach area (abdomen) or pain in the upper right side of the stomach area, sleepiness, or vomiting of blood [see Warnings and Precautions (5.2)].
Serious Symptomatic Bradycardia When Coadministered with Amiodarone
Advise patients to seek medical evaluation immediately for symptoms of bradycardia such as near-fainting or fainting, dizziness or lightheadedness, malaise, weakness, excessive tiredness, shortness of breath, chest pain, confusion, or memory problems [see Warnings and Precautions (5.3), Adverse Reactions (6.2), and Drug Interactions (7.3)].
Drug Interactions
Inform patients that VOSEVI may interact with other drugs. Advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products including St. John's wort [see Warnings and Precautions (5.3, 5.4) and Drug Interactions (7)].
Spl Unclassified Section
Manufactured and distributed by:Gilead Sciences, Inc.Foster City, CA 94404
VOSEVI, ATRIPLA, GENVOYA, HARVONI, and ODEFSEY are trademarks of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners.
© 2019 Gilead Sciences, Inc. All rights reserved.209195-GS-003
Patient Package Insert
| This Patient Information has been approved by the U.S. Food and Drug Administration. | Revised: 11/2019 | ||
| Patient InformationVOSEVI® (voh-SEV-ee) (sofosbuvir, velpatasvir, and voxilaprevir) tablets | |||
| What is the most important information I should know about VOSEVI? VOSEVI can cause serious side effects, including, Hepatitis B virus reactivation: Before starting treatment with VOSEVI, your healthcare provider will do blood tests to check for hepatitis B virus infection. If you have ever had hepatitis B virus infection, the hepatitis B virus could become active again during or after treatment of hepatitis C virus with VOSEVI. Hepatitis B virus becoming active again (called reactivation) may cause serious liver problems including liver failure and death. Your healthcare provider will monitor you if you are at risk for hepatitis B virus reactivation during treatment and after you stop taking VOSEVI. For more information about side effects, see the section "What are the possible side effects of VOSEVI?" | |||
| What is VOSEVI? VOSEVI is a prescription medicine used to treat adults with chronic (lasting a long time) hepatitis C virus (HCV) infection without cirrhosis or with compensated cirrhosis who have:
| |||
| Do not take VOSEVI: if you also take any medicines that contain rifampin (Rifater®, Rifamate®, Rimactane®, Rifadin®) | |||
Before taking VOSEVI, tell your healthcare provider about all your medical conditions, including if you:
Keep a list of your medicines to show your healthcare provider and pharmacist.
| |||
How should I take VOSEVI?
| |||
What are the possible side effects of VOSEVI?VOSEVI may cause serious side effects, including:
| |||
|
| ||
| |||
|
| ||
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |||
How should I store VOSEVI?
| |||
| General information about the safe and effective use of VOSEVI Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use VOSEVI for a condition for which it was not prescribed. Do not give VOSEVI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about VOSEVI that is written for health professionals. | |||
| What are the ingredients in VOSEVI?Active ingredients: sofosbuvir, velpatasvir, and voxilaprevir Inactive ingredients: colloidal silicon dioxide, copovidone, croscarmellose sodium, lactose monohydrate, magnesium stearate, and microcrystalline cellulose. The tablet film-coat contains: ferrosoferric oxide, iron oxide red, iron oxide yellow, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide. Manufactured and distributed by: Gilead Sciences, Inc., Foster City, CA 94404 For more information, call 1-800-445-3235 or go to www.VOSEVI.com. VOSEVI is a trademark of Gilead Sciences, Inc., or its related companies. All other trademarks referenced herein are the property of their respective owners. ©2019 Gilead Sciences, Inc. All rights reserved. 209195-GS-003 | |||