 Elbasvir-Grazoprevir Zepatier
Elbasvir-Grazoprevir Zepatier Glecaprevir-Pibrentasvir Mavyret
Glecaprevir-Pibrentasvir Mavyret Ledipasvir-Sofosbuvir Harvoni
Ledipasvir-Sofosbuvir Harvoni Ribavirin Copegus, Rebetol, Ribasphere
Ribavirin Copegus, Rebetol, Ribasphere Sofosbuvir Sovaldi
Sofosbuvir Sovaldi Sofosbuvir-Velpatasvir Epclusa
Sofosbuvir-Velpatasvir Epclusa Sofosbuvir-Velpatasvir-Voxilaprevir Vosevi
Sofosbuvir-Velpatasvir-Voxilaprevir VoseviBoxed Warning
WARNING: RISK OF HEPATITIS B VIRUS REACTIVATION IN PATIENTS COINFECTED WITH HCV AND HBV
Test all patients for evidence of current or prior hepatitis B virus (HBV) infection before initiating treatment with MAVYRET. HBV reactivation has been reported in HCV/HBV coinfected patients who were undergoing or had completed treatment with HCV direct-acting antivirals and were not receiving HBV antiviral therapy. Some cases have resulted in fulminant hepatitis, hepatic failure, and death. Monitor HCV/HBV coinfected patients for hepatitis flare or HBV reactivation during HCV treatment and post-treatment follow-up. Initiate appropriate patient management for HBV infection as clinically indicated [see Warnings and Precautions (5.1)].
1. Indications and Usage
MAVYRET is indicated for the treatment of adult and pediatric patients 3 years and older with chronic hepatitis C virus (HCV) genotype 1, 2, 3, 4, 5 or 6 infection without cirrhosis or with compensated cirrhosis (Child-Pugh A).
MAVYRET is indicated for the treatment of adult and pediatric patients 3 years and older with HCV genotype 1 infection, who previously have been treated with a regimen containing an HCV NS5A inhibitor or an NS3/4A protease inhibitor (PI), but not both [see Dosage and Administration ( 2.2 ) and Clinical Studies ( 14 )].
2. Dosage and Administration
2.1 Testing Prior to the Initiation of Therapy
Test all patients for evidence of current or prior HBV infection by measuring hepatitis B surface antigen (HBsAg) and hepatitis B core antibody (anti-HBc) before initiating HCV treatment with MAVYRET [see Warnings and Precautions ( 5.1 ) ].
2.2 Recommended Treatment Duration in Patients 3 Years and Older
Tables 1 and 2 provide the recommended MAVYRET treatment duration based on the patient population in HCV mono-infected and HCV/HIV-1 co-infected patients with compensated liver disease (with or without cirrhosis) and with or without renal impairment including patients receiving dialysis [see Contraindications ( 4 ) and Clinical Studies ( 14 )]. Refer to Drug Interactions ( 7 ) for dosage recommendations for concomitant HIV-1 antiviral drugs.
| Table 1. Recommended Duration for Treatment-Naïve Patients | ||
| HCV Genotype | Treatment Duration | |
| No Cirrhosis | Compensated Cirrhosis(Child-Pugh A) | |
| 1, 2, 3, 4, 5, or 6 | 8 weeks | 8 weeks |
| Table 2. Recommended Duration for Treatment-Experienced Patients | |||
| Treatment Duration | |||
| HCV Genotype | Patients PreviouslyTreated with aRegimen Containing: | No Cirrhosis | CompensatedCirrhosis(Child-Pugh A) |
| 1 | An NS5A inhibitor1 without prior treatment with an NS3/4A protease inhibitor (PI) | 16 weeks | 16 weeks |
| An NS3/4A PI2 without prior treatment with an NS5A inhibitor | 12 weeks | 12 weeks | |
| 1, 2, 4, 5, or 6 | PRS3 | 8 weeks | 12 weeks |
| 3 | PRS3 | 16 weeks | 16 weeks |
| |||
2.3 Recommended Dosage in Adults
MAVYRET tablets are a fixed combination drug product containing glecaprevir 100 mg and pibrentasvir 40 mg in each tablet.
The recommended oral dosage of MAVYRET in adults is 3 tablets taken at the same time once daily with food (total daily dose: glecaprevir 300 mg and pibrentasvir 120 mg) [see Clinical Pharmacology ( 12.3 )].
2.4 Recommended Dosage in Pediatric Patients 3 Years of Age and Older
The recommended dosage of MAVYRET in pediatric patients 3 to less than 12 years of age is based on weight. MAVYRET oral pellets are recommended for pediatric patients 3 to less than 12 years old weighing less than 45 kg. MAVYRET oral pellets in packets are a fixed combination drug product containing glecaprevir 50 mg and pibrentasvir 20 mg in each packet.
The recommended dosage of MAVYRET in pediatric patients 12 years of age and older, or in pediatric patients weighing at least 45 kg, is three tablets taken at the same time once daily with food (total daily dose: glecaprevir 300 mg and pibrentasvir 120 mg).
The dosages for pediatric patients are shown in Table 3.
| Table 3: Recommended Dosage in Pediatric Patients 3 Years of Age and Older | ||
| Body Weight (kg) or Age (yrs) | Daily Dose of glecaprevir/pibrentasvir | Dosing of MAVYRET |
| Less than 20 kg | 150 mg/60 mg per day | Three 50 mg/20 mg packets of oral pellets once daily |
| 20 kg to less than 30 kg | 200 mg/80 mg per day | Four 50 mg/20 mg packets of oral pellets once daily |
| 30 kg to less than 45 kg | 250 mg/100 mg per day | Five 50 mg/20 mg packets of oral pellets once daily |
| 45 kg and greater OR 12 years of age and older | 300 mg/120 mg per day | Three 100 mg/40 mg tablets once daily1 (see Recommended Dosage in Adults) |
| 1 Pediatric patients weighing 45 kg and greater who are unable to swallow tablets may take six 50 mg/20 mg packets of oral pellets once daily. Dosing with oral pellets has not been studied for pediatric patients weighing greater than 45 kg [see Clinical Pharmacology ( 12.3 ) ]. | ||
2.5 Preparation and Administration of Oral Pellets
See the MAVYRET oral pellets full Instructions for Use for details on the preparation and administration.
- The oral pellets should be taken together, with food, once daily. In addition, the oral pellets for the total daily dose should be sprinkled on a small amount of soft food with a low water content that will stick to a spoon and should be swallowed without chewing (e.g., peanut butter, chocolate hazelnut spread, cream cheese, thick jam, or Greek yogurt).
- The entire mixture of food and oral pellets should be swallowed within 15 minutes of preparation; the oral pellets should not be crushed or chewed.
- Liquids or foods that would drip or slide off the spoon are not recommended as the drug may dissolve quickly and become less effective.
2.6 Liver or Kidney Transplant Recipients
MAVYRET is recommended for 12 weeks in patients 3 years and older who are liver or kidney transplant recipients. A 16-week treatment duration is recommended in genotype 1-infected patients who are NS5A inhibitor-experienced without prior treatment with an NS3/4A protease inhibitor or in genotype 3-infected patients who are PRS treatment-experienced [see Clinical Studies ( 14.8 )].
3. Dosage Forms and Strengths
MAVYRET is available as tablets or pellets for oral use.
- Tablets: pink, oblong-shaped, film-coated, and debossed with “NXT” on one side. Each tablet contains 100 mg glecaprevir and 40 mg of pibrentasvir.
- Oral pellets: pink and yellow coated pellets in unit-dose packets. Each packet contains 50 mg glecaprevir and 20 mg pibrentasvir.
4. Contraindications
- MAVYRET is contraindicated in patients with moderate or severe hepatic impairment (Child-Pugh B or C) or those with any history of prior hepatic decompensation [see Warnings and Precautions ( 5.2 ), Use in Specific Populations ( 8.7 ) and Clinical Pharmacology ( 12.3 )].
- MAVYRET is contraindicated with atazanavir or rifampin [see Drug Interaction ( 7.3 ) and Clinical Pharmacology ( 12.3 )].
5. Warnings and Precautions
5.1 Risk of Hepatitis B Virus Reactivation in Patients Coinfected with HCV and HBV
Hepatitis B virus (HBV) reactivation has been reported in HCV/HBV coinfected patients who were undergoing or had completed treatment with HCV direct-acting antivirals, and who were not receiving HBV antiviral therapy. Some cases have resulted in fulminant hepatitis, hepatic failure and death. Cases have been reported in patients who are HBsAg positive and also in patients with serologic evidence of resolved HBV infection (i.e., HBsAg negative and anti-HBc positive). HBV reactivation has also been reported in patients receiving certain immunosuppressant or chemotherapeutic agents; the risk of HBV reactivation associated with treatment with HCV direct-acting antivirals may be increased in these patients.
HBV reactivation is characterized as an abrupt increase in HBV replication manifesting as a rapid increase in serum HBV DNA level. In patients with resolved HBV infection reappearance of HBsAg can occur. Reactivation of HBV replication may be accompanied by hepatitis, i.e., increase in aminotransferase levels and, in severe cases, increases in bilirubin levels, liver failure, and death can occur.
Test all patients for evidence of current or prior HBV infection by measuring HBsAg and anti- HBc before initiating HCV treatment with MAVYRET. In patients with serologic evidence of HBV infection, monitor for clinical and laboratory signs of hepatitis flare or HBV reactivation during HCV treatment with MAVYRET and during post-treatment follow-up. Initiate appropriate patient management for HBV infection as clinically indicated.
5.2 Risk of Hepatic Decompensation/Failure in Patients with Evidence of Advanced Liver Disease
Postmarketing cases of hepatic decompensation/failure, including those with fatal outcomes, have been reported in patients treated with HCV NS3/4A protease inhibitor-containing regimens, including MAVYRET. Because these events are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
The majority of patients with severe outcomes had evidence of advanced liver disease with moderate or severe hepatic impairment (Child-Pugh B or C) prior to initiating therapy with MAVYRET, including some patients reported as having compensated cirrhosis with mild liver impairment (Child-Pugh A) at baseline but with a prior decompensation event (i.e., prior history of ascites, variceal bleeding, encephalopathy). Rare cases of hepatic decompensation/failure were reported in patients without cirrhosis or with compensated cirrhosis (Child-Pugh A); many of these patients had evidence of portal hypertension. Events also occurred in patients taking a concomitant medication not recommended for coadministration, or in patients with confounding factors such as serious liver-related medical or surgical comorbidities. Cases typically occurred within the first 4 weeks of treatment (median of 27 days).
In patients with compensated cirrhosis (Child-Pugh A) or evidence of advanced liver disease such as portal hypertension, perform hepatic laboratory testing as clinically indicated; and monitor for signs and symptoms of hepatic decompensation such as the presence of jaundice, ascites, hepatic encephalopathy, and variceal hemorrhage. Discontinue MAVYRET in patients who develop evidence of hepatic decompensation/failure.
MAVYRET is contraindicated in patients with moderate to severe hepatic impairment (Child-Pugh B or C) or those with any history of prior hepatic decompensation [see Contraindications ( 4 ), Adverse Reactions ( 6.1 ), Use in Specific Populations ( 8.7 ), and Clinical Pharmacology ( 12.3 )].
5.3 Risk of Reduced Therapeutic Effect Due to Concomitant Use of MAVYRET with Carbamazepine, Efavirenz Containing Regimens, or St. John’s Wort
Carbamazepine, efavirenz, and St. John’s wort may significantly decrease plasma concentrations of glecaprevir and pibrentasvir, leading to reduced therapeutic effect of MAVYRET. The use of these agents with MAVYRET is not recommended.
6. Adverse Reactions
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of MAVYRET cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Overall Adverse Reactions in Subjects without Cirrhosis or with Compensated Cirrhosis (Child-Pugh A)
The adverse reactions data for MAVYRET in subjects without cirrhosis or with compensated cirrhosis (Child-Pugh A) were derived from nine registrational Phase 2 and 3 trials which evaluated approximately 2,300 adults infected with genotype 1, 2, 3, 4, 5, or 6 HCV who received MAVYRET for 8, 12 or 16 weeks [see Clinical Studies ( 14 ) ].
The overall proportion of subjects who permanently discontinued treatment due to adverse reactions was 0.1% for subjects who received MAVYRET for 8, 12 or 16 weeks.
The most common adverse reactions, all grades, observed in greater than or equal to 5% of subjects receiving 8, 12, or 16 weeks of treatment with MAVYRET were headache (13%), fatigue (11%), and nausea (8%). In subjects receiving MAVYRET who experienced adverse reactions, 80% had an adverse reaction of mild severity (Grade 1). One subject experienced a serious adverse reaction.
Adverse reactions (type and severity) were similar for subjects receiving MAVYRET for 8, 12 or 16 weeks. The type and severity of adverse reactions in subjects with compensated cirrhosis (Child-Pugh A) were similar to those seen in subjects without cirrhosis.
Adverse Reactions in Subjects without Cirrhosis
ENDURANCE-2
Among 302 treatment-naïve or PRS treatment-experienced, HCV genotype 2-infected adults without cirrhosis enrolled in ENDURANCE-2, adverse reactions (all intensity) occurring in at least 5% of subjects treated with MAVYRET for 12 weeks are presented in Table 4. In subjects treated with MAVYRET for 12 weeks, 32% reported an adverse reaction, of which 98% had adverse reactions of mild or moderate severity. No subjects treated with MAVYRET or placebo in ENDURANCE-2 permanently discontinued treatment due to an adverse drug reaction.
| Table 4. Adverse Reactions Reported in ≥5% of Treatment-Naïve and PRS-Experienced Adults without Cirrhosis Receiving MAVYRET for 12 Weeks in ENDURANCE-2 | ||
| Adverse Reaction | MAVYRET12 Weeks(N = 202)% | Placebo12 Weeks(N = 100)% |
| Headache | 9 | 6 |
| Nausea | 6 | 2 |
| Diarrhea | 5 | 2 |
ENDURANCE-3
Among 505 treatment-naïve, HCV genotype 3-infected adults without cirrhosis enrolled in ENDURANCE-3, adverse reactions (all intensity) occurring in at least 5% of subjects treated with MAVYRET for 8 or 12 weeks are presented in Table 5. In subjects treated with MAVYRET, 45% reported an adverse reaction, of which 99% had adverse reactions of mild or moderate severity. The proportion of subjects who permanently discontinued treatment due to adverse reactions was 0%, < 1% and 1% for the MAVYRET 8-week arm, MAVYRET 12 week arm and DCV + SOF arm, respectively.
| Table 5. Adverse Reactions Reported in ≥5% of Treatment-Naïve Adults without Cirrhosis Receiving MAVYRET for 8 Weeks or 12 Weeks in ENDURANCE-3 | |||
| Adverse Reaction | MAVYRET*8 Weeks(N = 157)% | MAVYRET 12 Weeks (N = 233) % | DCV1 + SOF2 12 Weeks (N = 115) % |
| Headache | 16 | 17 | 15 |
| Fatigue | 11 | 14 | 12 |
| Nausea | 9 | 12 | 12 |
| Diarrhea | 7 | 3 | 3 |
| 1 DCV=daclatasvir 2 SOF=sofosbuvir * The 8-week arm was a non-randomized treatment arm. | |||
Adverse Reactions in Subjects with Compensated Cirrhosis (Child-Pugh A)
The safety of MAVYRET in HCV GT 1, 2, 3, 4, 5, or 6 subjects with compensated cirrhosis is based on data from 288 adults from the Phase 2/3 registrational trials treated with MAVYRET for 12 or more weeks and 343 adults from EXPEDITION-8 treated with MAVYRET for 8 weeks. The adverse reactions observed were generally consistent with those observed in clinical studies of MAVYRET in non-cirrhotic subjects [see Clinical Studies ( 14 )].
In the Phase 2/3 registrational trials, the adverse reactions reported in greater than or equal to 5% of compensated cirrhotic subjects (n=288) treated across all durations of MAVYRET were fatigue (15%), headache (14%), nausea (8%), diarrhea (6%), and pruritus (6%). In EXPEDITION-8, the adverse reactions reported in greater than or equal to 5% of compensated cirrhotic subjects (n=343) were fatigue (8%), pruritus (7%), and headache (6%). No subjects with compensated cirrhosis in the Phase 2/3 registrational trials (without severe renal impairment) or in EXPEDITION-8 discontinued treatment with MAVYRET due to an adverse reaction.
Adverse Reactions in Subjects with Severe Renal Impairment Including Those on Dialysis
The safety of MAVYRET in subjects with chronic kidney disease (Stage 4 or Stage 5 including subjects on dialysis) with genotypes 1, 2, 3, 4, 5 or 6 chronic HCV infection without cirrhosis or with compensated cirrhosis (Child-Pugh A) was assessed in 104 adults (EXPEDITION-4) who received MAVYRET for 12 weeks. The most common adverse reactions observed in greater than or equal to 5% of subjects receiving 12 weeks of treatment with MAVYRET were pruritus (17%), fatigue (12%), nausea (9%), asthenia (7%), and headache (6%). In subjects treated with MAVYRET who reported an adverse reaction, 90% had adverse reactions of mild or moderate severity (Grade 1 or 2). The proportion of subjects who permanently discontinued treatment due to adverse reactions was 2%.
Adverse Reactions in HCV/HIV-1 Co-infected Subjects
The safety of MAVYRET in subjects with HIV-1 co-infection with genotypes 1, 2, 3, 4 or 6 chronic HCV infection without cirrhosis or with compensated cirrhosis (Child-Pugh A) was assessed in 153 adults (EXPEDITION-2) who received MAVYRET for 8 or 12 weeks. Thirty-three subjects with HIV-1 coinfection also received 8 or 12 weeks of therapy in ENDURANCE-1.
The overall safety profile in HCV/HIV-1 co-infected subjects (ENDURANCE-1 and EXPEDITION-2) was similar to that observed in HCV mono-infected subjects. Adverse reactions observed in greater than or equal to 5% of subjects receiving MAVYRET in EXPEDITION-2 for 8 or 12 weeks were fatigue (10%), nausea (8%), and headache (5%).
Adverse Reactions in Subjects with Liver or Kidney Transplant
The safety of MAVYRET was assessed in 100 adult post-liver or -kidney transplant recipients with genotypes 1, 2, 3, 4, or 6 chronic HCV infection without cirrhosis (MAGELLAN-2). The overall safety profile in transplant recipients was similar to that observed in subjects in the Phase 2 and 3 studies, without a history of transplantation. Adverse reactions observed in greater than or equal to 5% of subjects receiving MAVYRET for 12 weeks were headache (17%), fatigue (16%), nausea (8%) and pruritus (7%). In subjects treated with MAVYRET who reported an adverse reaction, 81% had adverse reactions of mild severity. Two percent of subjects experienced a serious adverse reaction, and no subjects permanently discontinued treatment due to adverse reactions.
Adverse Reactions in People Who Inject Drugs (PWID) and those on Medication-Assisted Treatment (MAT) for Opioid Use Disorder
The safety of MAVYRET in PWID with HCV GT 1, 2, 3, 4, 5, or 6 infection is based on data from adults and adolescents in Phase 2 and 3 trials in which 62 subjects identified as current/recent PWID (defined as self-reported injection drug use within the last 12 months prior to starting MAVYRET) and 3,282 subjects reported no injection drug use (non-PWID).
Among current/recent PWID, adverse reactions observed in greater than or equal to 5% of subjects were fatigue (16%), headache (13%), diarrhea (6%), and nausea (6%). Among non-PWID subjects, adverse reactions observed in greater than or equal to 5% were headache (7%) and fatigue (6%). Serious adverse reactions and/or adverse reactions leading to treatment discontinuation occurred in one current/recent PWID subject (2%) compared to less than 1% in non-PWID subjects [see Use in Specific Populations ( 8.8 ) and Clinical Studies ( 14.9 ) ].
Among 225 subjects reporting concomitant use of MAT for opioid use disorder, adverse reactions observed in greater than or equal to 5% were headache (15%), fatigue (12%), nausea (11%), and diarrhea (6%). Among 4,098 subjects who were not on MAT, adverse reactions observed in greater than or equal to 5% were headache (9%), fatigue (8%), and nausea (5%). Serious adverse reactions and/or adverse reactions leading to treatment discontinuation were not observed among subjects on MAT and were experienced by less than 1% of subjects not on MAT [see Use in Specific Populations ( 8.8 ) and Clinical Studies ( 14.9 ) ].
Adverse Reactions in Pediatric Subjects 3 Years and Older
The safety of MAVYRET in HCV GT1, 2, 3, or 4 infected adolescents is based on data from a Phase 2/3 open-label trial in 47 subjects aged 12 years to less than 18 years without cirrhosis treated with MAVYRET for 8 or 16 weeks (DORA-Part 1). The adverse reactions observed in subjects 12 years to less than 18 years of age were consistent with those observed in clinical trials of MAVYRET in adults. The only adverse reaction observed in greater than or equal to 5% of subjects receiving MAVYRET in DORA Part 1 was fatigue (6%). No subjects discontinued or interrupted treatment with MAVYRET due to an adverse reaction.
The safety of MAVYRET in HCV GT 1, 2, 3, or 4 infected pediatric subjects aged 3 years to less than 12 years is based on data from a Phase 2/3 open-label trial in 80 subjects aged 3 to less than 12 years without cirrhosis treated with weight-based MAVYRET oral pellets in packets for 8, 12 or 16 weeks (DORA-Part 2). The adverse reactions observed in subjects 3 years to less than 12 years of age were consistent with those observed in clinical trials of MAVYRET in adults with the exception of vomiting (occurring at 8%), rash, and abdominal pain upper (each occurring at 4%) which were observed more frequently in pediatric subjects less than 12 years of age compared to adults. Other adverse reactions observed in greater than or equal to 5% of subjects receiving MAVYRET in DORA-Part 2 include fatigue and headache, each occurring at 8%. One subject discontinued treatment due to an adverse reaction of erythematous rash (Grade 3). All other adverse reactions were Grade 1 or 2 and no subjects interrupted treatment due to an adverse reaction [see Use in Specific Populations ( 8.4 ), Clinical Studies ( 14.10 ) ].
Laboratory Abnormalities
Serum bilirubin elevations
Elevations of total bilirubin at least 2 times the upper limit of normal occurred in 3.5% of adult subjects treated with MAVYRET versus 0% in placebo; these elevations were observed in 1.2% of adult subjects across the Phase 2 and 3 trials.
In adult subjects with compensated cirrhosis (Child-Pugh A), 17% experienced early, transient post-baseline elevations of bilirubin above the upper limit of normal. These bilirubin elevations were typically less than two times the upper limit of normal, generally occurred within the first 2 weeks of treatment and resolved with continued treatment. The subjects with compensated cirrhosis and bilirubin elevations did not have concurrent increases in ALT or AST, or signs of liver decompensation or failure, and these laboratory events did not lead to treatment discontinuation. MAVYRET inhibits OATP1B1/3 and is a weak inhibitor of UGT1A1 and may have the potential to impact bilirubin transport and metabolism, including direct and indirect bilirubin. Few subjects experienced jaundice or ocular icterus and total bilirubin levels decreased after completing MAVYRET.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of MAVYRET. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Skin and Subcutaneous Tissue Disorders: Angioedema
Hepatobiliary Disorders: Hepatic decompensation, hepatic failure [see Warnings and Precautions ( 5.2 )].
7. Drug Interactions
7.1 Mechanisms for the Potential Effect of MAVYRET on Other Drugs
Glecaprevir and pibrentasvir are inhibitors of P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), and organic anion transporting polypeptide (OATP) 1B1/3. Coadministration with MAVYRET may increase plasma concentration of drugs that are substrates of P-gp, BCRP, OATP1B1 or OATP1B3. Glecaprevir and pibrentasvir are weak inhibitors of cytochrome P450 (CYP) 3A, CYP1A2, and uridine glucuronosyltransferase (UGT) 1A1.
7.2 Mechanisms for the Potential Effect of Other Drugs on MAVYRET
Glecaprevir and pibrentasvir are substrates of P-gp and/or BCRP. Glecaprevir is a substrate of OATP1B1/3. Coadministration of MAVYRET with drugs that inhibit hepatic P-gp, BCRP, or OATP1B1/3 may increase the plasma concentrations of glecaprevir and/or pibrentasvir.
Coadministration of MAVYRET with drugs that induce P-gp/CYP3A may decrease glecaprevir and pibrentasvir plasma concentrations.
Carbamazepine, phenytoin, efavirenz, and St. John’s wort may significantly decrease plasma concentrations of glecaprevir and pibrentasvir, leading to reduced therapeutic effect of MAVYRET. The use of these agents with MAVYRET is not recommended [see Warnings and Precautions ( 5.3 ) and Clinical Pharmacology ( 12.3 )].
7.3 Established and Other Potential Drug Interactions
Clearance of HCV infection with direct-acting antivirals may lead to changes in hepatic function, which may impact the safe and effective use of concomitant medications. For example, altered blood glucose control resulting in serious symptomatic hypoglycemia has been reported in diabetic patients in postmarketing case reports and published epidemiological studies. Management of hypoglycemia in these cases required either discontinuation or dose modification of concomitant medications used for diabetes treatment.
Frequent monitoring of relevant laboratory parameters (e.g. International Normalized Ratio [INR] in patients taking warfarin, blood glucose levels in diabetic patients) or drug concentrations of concomitant medications such as CYP P450 substrates with a narrow therapeutic index (e.g. certain immunosuppressants) is recommended to ensure safe and effective use. Dose adjustments of concomitant medications may be necessary.
Table 6 provides the effect of MAVYRET on concentrations of coadministered drugs and the effect of coadministered drugs on glecaprevir and pibrentasvir [see Contraindications ( 4 ) , Warnings and Precautions ( 5.3 ) , and Clinical Pharmacology ( 12.3 ) ]. All interaction studies were performed in adults.
| Table 6. Potentially Significant Drug Interactions Identified in Drug Interaction Studies | ||
| ConcomitantDrug Class:Drug Name | Effect onConcentration | Clinical Comments |
| Antiarrhythmics: | ||
| Digoxin | ↑ digoxin | Measure serum digoxin concentrations before initiating MAVYRET. Reduce digoxin concentrations by decreasing the dose by approximately 50% or by modifying the dosing frequency and continue monitoring. |
| Anticoagulants: | ||
| Dabigatran etexilate | ↑ dabigatran | If MAVYRET and dabigatran etexilate are coadministered, refer to the dabigatran etexilate prescribing information for dabigatran etexilate dose modifications in combination with P-gp inhibitors in the setting of renal impairment. |
| Anticonvulsants: | ||
| Carbamazepine | ↓ glecaprevir ↓ pibrentasvir | Coadministration may lead to reduced therapeutic effect of MAVYRET and is not recommended. |
| Antimycobacterials: | ||
| Rifampin | ↓ glecaprevir ↓ pibrentasvir | Coadministration is contraindicated because of potential loss of therapeutic effect [see Contraindications ( 4 ) ]. |
| Ethinyl Estradiol-Containing Products: | ||
| Ethinyl estradiol-containing medications such as combined oral contraceptives | ↔ glecaprevir ↔ pibrentasvir | MAVYRET may be used with products containing 20 mcg or less of ethinyl estradiol. Coadministration of MAVYRET with products containing more than 20 mcg of ethinyl estradiol may increase the risk of ALT elevations and is not recommended. |
| Herbal Products: | ||
| St. John’s wort (hypericum perforatum) | ↓ glecaprevir ↓ pibrentasvir | Coadministration may lead to reduced therapeutic effect of MAVYRET and is not recommended. |
| HIV-Antiviral Agents: | ||
| Atazanavir | ↑ glecaprevir ↑ pibrentasvir | Coadministration is contraindicated due to increased risk of ALT elevations [see Contraindications ( 4 ) ]. |
| Darunavir Lopinavir Ritonavir | ↑ glecaprevir ↑ pibrentasvir | Coadministration is not recommended. |
| Efavirenz | ↓ glecaprevir ↓ pibrentasvir | Coadministration may lead to reduced therapeutic effect of MAVYRET and is not recommended. |
| HMG-CoA Reductase Inhibitors: | ||
| Atorvastatin Lovastatin Simvastatin | ↑ atorvastatin ↑ lovastatin ↑ simvastatin | Coadministration may increase the concentration of atorvastatin, lovastatin, and simvastatin. Increased statin concentrations may increase the risk of myopathy, including rhabdomyolysis. Coadministration with these statins is not recommended. |
| Pravastatin | ↑ pravastatin | Coadministration may increase the concentration of pravastatin. Increased statin concentrations may increase the risk of myopathy, including rhabdomyolysis. Reduce pravastatin dose by 50% when coadministered with MAVYRET. |
| Rosuvastatin | ↑ rosuvastatin | Coadministration may significantly increase the concentration of rosuvastatin. Increased statin concentrations may increase the risk of myopathy, including rhabdomyolysis. Rosuvastatin may be administered with MAVYRET at a dose that does not exceed 10 mg. |
| Fluvastatin Pitavastatin | ↑ fluvastatin ↑ pitavastatin | Coadministration may increase the concentrations of fluvastatin and pitavastatin. Increased statin concentrations may increase the risk of myopathy, including rhabdomyolysis. Use the lowest approved dose of fluvastatin or pitavastatin. If higher doses are needed, use the lowest necessary statin dose based on a risk/benefit assessment. |
| Immunosuppressants: | ||
| Cyclosporine | ↑ glecaprevir ↑ pibrentasvir | MAVYRET is not recommended for use in patients requiring stable cyclosporine doses > 100 mg per day. |
| See Clinical Pharmacology, Tables 10 and 11. ↑= increase; ↓= decrease; ↔ = no effect | ||
7.4 Medication-Assisted Treatment (MAT) for Opioid Use Disorder
No buprenorphine/naloxone or methadone dosage adjustment is required when used concomitantly with MAVYRET. There is insufficient information to make a recommendation regarding the concomitant use of naltrexone with MAVYRET.
7.5 Drugs with No Observed Clinically Significant Interactions with MAVYRET
No dose adjustment is required when MAVYRET is coadministered with the following medications: abacavir, amlodipine, caffeine, dextromethorphan, dolutegravir, elvitegravir/ cobicistat, emtricitabine, ethinyl estradiol of 20 mcg or less, felodipine, lamivudine, lamotrigine, losartan, midazolam, norethindrone or other progestin-only contraceptives, omeprazole, raltegravir, rilpivirine, sofosbuvir, tacrolimus, tenofovir alafenamide, tenofovir disoproxil fumarate, tolbutamide, and valsartan.
8. Use in Specific Populations
8.1 Pregnancy
Risk Summary
No adequate human data are available to establish whether or not MAVYRET poses a risk to pregnancy outcomes. In animal reproduction studies, no adverse developmental effects were observed when the components of MAVYRET were administered separately during organogenesis at exposures up to 53 times (rats; glecaprevir) or 51 and 1.5 times (mice and rabbits, respectively; pibrentasvir) the human exposures at the recommended dose of MAVYRET (see Data ). No definitive conclusions regarding potential developmental effects of glecaprevir could be made in rabbits, since the highest achieved glecaprevir exposure in this species was only 7% (0.07 times) of the human exposure at the recommended dose. There were no effects with either compound in rodent pre/post-natal developmental studies in which maternal systemic exposures (AUC) to glecaprevir and pibrentasvir were approximately 47 and 74 times, respectively, the exposure in humans at the recommended dose (see Data ).
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Glecaprevir
Glecaprevir was administered orally to pregnant rats (up to 120 mg/kg/day) and rabbits (up to 60 mg/kg/day) during the period of organogenesis (gestation days (GD) 6 to 18, and GD 7 to 19, respectively). No adverse embryo-fetal effects were observed in rats at dose levels up to 120 mg/kg/day (53 times the exposures in humans at the recommended human dose (RHD)). In rabbits, the highest glecaprevir exposure achieved was 7% (0.07 times) of the exposure in humans at RHD. As such, data in rabbits during organogenesis are not available for glecaprevir systemic exposures at or above the exposures in humans at the RHD.
In the pre/post-natal developmental study in rats, glecaprevir was administered orally (up to 120 mg/kg/day) from GD 6 to lactation day 20. No effects were observed at maternal exposures 47 times the exposures in humans at the RHD.
Pibrentasvir
Pibrentasvir was administered orally to pregnant mice and rabbits (up to 100 mg/kg/day) during the period of organogenesis (GD 6 to 15, and GD 7 to 19, respectively). No adverse embryo-fetal effects were observed at any studied dose level in either species. The systemic exposures at the highest doses were 51 times (mice) and 1.5 times (rabbits) the exposures in humans at the RHD.
In the pre/post-natal developmental study in mice, pibrentasvir was administered orally (up to 100 mg/kg/day) from GD 6 to lactation day 20. No effects were observed at maternal exposures approximately 74 times the exposures in humans at the RHD.
8.2 Lactation
Risk Summary
It is not known whether the components of MAVYRET are excreted in human breast milk, affect human milk production, or have effects on the breastfed infant. When administered to lactating rodents, the components of MAVYRET were present in milk, without effect on growth and development observed in the nursing pups (see Data ).
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for MAVYRET and any potential adverse effects on the breastfed child from MAVYRET or from the underlying maternal condition.
Data
No significant effects of glecaprevir or pibrentasvir on growth and post-natal development were observed in nursing pups at the highest doses tested (120 mg/kg/day for glecaprevir and 100 mg/kg/day for pibrentasvir). Maternal systemic exposure (AUC) to glecaprevir and pibrentasvir was approximately 47 or 74 times the exposure in humans at the RHD. Systemic exposure in nursing pups on post-natal day 14 was approximately 0.6 to 2.2 % of the maternal exposure for glecaprevir and approximately one quarter to one third of the maternal exposure for pibrentasvir.
Glecaprevir or pibrentasvir was administered (single dose; 5 mg/kg oral) to lactating rats, 8 to 12 days post parturition. Glecaprevir in milk was 13 times lower than in plasma and pibrentasvir in milk was 1.5 times higher than in plasma. Parent drug (glecaprevir or pibrentasvir) represented the majority (>96%) of the total drug-related material in milk.
8.4 Pediatric Use
No dosage adjustment of MAVYRET is required in pediatric patients 12 years and older or weighing at least 45 kg. The recommended dosage of MAVYRET in pediatric patients 3 to less than 12 years of age is based on weight [see Dosage and Administration ( 2.2 , 2.4 ) , Clinical Pharmacology ( 12.3 ) and Clinical Studies ( 14.10 ) ].
The safety, efficacy, and pharmacokinetics of MAVYRET in HCV GT1, 2, 3, or 4 infected pediatric patients 3 years and older is based on data from an open-label trial in 127 subjects without cirrhosis aged 3 years to less than 18 years who were either treatment-naïve (n=114) or treatment-experienced (n=13) and received MAVYRET for 8, 12 or 16 weeks (DORA-Part 1 and Part 2).
The adverse reactions observed in subjects 3 years to less than 18 years of age were consistent with those observed in clinical trials of MAVYRET in adults with the exception of vomiting, rash and abdominal pain upper which were observed more frequently in pediatric subjects less than 12 years of age compared to adults [see Adverse Reactions ( 6.1 ) ].
The efficacy results observed in this trial were consistent with those observed in clinical trials of MAVYRET in adults [see Clinical Studies ( 14.10 ) ].
In pediatric patients with cirrhosis, history of a kidney and/or liver transplant, or HCV GT5 or 6 infection, the safety and efficacy of MAVYRET are supported by the comparable glecaprevir and pibrentasvir exposures observed between pediatric subjects and adults [see Clinical Pharmacology ( 12.3 ) ].
The safety and effectiveness of MAVYRET in children less than 3 years of age have not been studied.
8.5 Geriatric Use
In clinical trials of MAVYRET, 328 subjects were age 65 years and over (14% of the total number of subjects in the Phase 2 and 3 clinical trials) and 47 subjects were age 75 and over (2%). No overall differences in safety or effectiveness were observed between these subjects and younger subjects, and other reported clinical experience has not identified differences in responses between the elderly and younger subjects. No dosage adjustment of MAVYRET is warranted in geriatric patients [see Clinical Pharmacology ( 12.3 ) ].
8.7 Hepatic Impairment
No dosage adjustment of MAVYRET is required in patients with mild hepatic impairment (Child-Pugh A). MAVYRET has not been evaluated and is contraindicated in HCV-infected patients with moderate or severe hepatic impairment (Child-Pugh B or Child-Pugh C) or those with any history of prior hepatic decompensation [see Contraindications ( 4 )]. Postmarketing cases of hepatic decompensation/failure have been reported in these patients [see Warnings and Precautions ( 5.2 )]. Higher exposures of both glecaprevir and pibrentasvir occur in subjects with severe hepatic impairment (Child-Pugh C) [see Clinical Pharmacology ( 12.3 )].
8.8 People Who Inject Drugs (PWID) and those on Medication-Assisted Treatment (MAT) for Opioid Use Disorder
No dosage adjustment of MAVYRET is required in PWID or those who are on MAT for opioid use disorder. In clinical trials of MAVYRET, the safety and efficacy were similar between subjects who self-identified as current/recent PWID, those who were former PWID, and those who did not report history of injection drug use. The safety and efficacy of MAVYRET were also similar between subjects who reported concomitant MAT for opioid use disorder and those who did not report concomitant MAT [see Adverse Reactions ( 6.1 ), Drug Interactions ( 7.4 ) and Clinical Studies ( 14.9 )].
10. Overdosage
11. Description
MAVYRET contains glecaprevir, a HCV NS3/4A PI, and pibrentasvir a HCV NS5A inhibitor. MAVYRET is available as a fixed dose combination tablet or coated pellets in unit-dose packets for oral administration.
Glecaprevir/Pibrentasvir Film-Coated Immediate Release Tablets
Each tablet contains 100 mg of glecaprevir and 40 mg of pibrentasvir. Glecaprevir and pibrentasvir are presented as a co-formulated, fixed-dose combination, immediate release bilayer tablet.
The tablet contains the following inactive ingredients: colloidal silicon dioxide, copovidone (type K 28), croscarmellose sodium, hypromellose 2910, iron oxide red, lactose monohydrate, polyethylene glycol 3350, propylene glycol monocaprylate (type II), sodium stearyl fumarate, titanium dioxide, and vitamin E (tocopherol) polyethylene glycol succinate.
The tablets do not contain gluten.
Glecaprevir/Pibrentasvir Coated Oral Pellets
MAVYRET oral pellets are small, pink and yellow and supplied in unit-dose packets for oral administration. Each unit-dose of MAVYRET oral pellets in packets contains 50 mg glecaprevir and 20 mg pibrentasvir and the following inactive ingredients: colloidal silicon dioxide, copovidone (type K 28), croscarmellose sodium, hypromellose 2910, iron oxide red, iron oxide yellow, lactose monohydrate, polyethylene glycol/macrogol 3350, propylene glycol monocaprylate (type II), sodium stearyl fumarate, titanium dioxide, vitamin E (tocopherol) polyethylene glycol succinate.
The oral pellets do not contain gluten.
Glecaprevir drug substance:
The chemical name of glecaprevir is (3aR,7S,10S,12R,21E,24aR)-7-tert-butyl-N-{(1R,2R)-2-(difluoromethyl)-1-[(1-methylcyclopropane-1-sulfonyl)carbamoyl]cyclopropyl}-20,20-difluoro-5,8-dioxo-2,3,3a,5,6,7,8,11,12,20,23,24a-dodecahydro-1H,10H-9,12-methanocyclopenta[18,19][1,10,17,3,6]trioxadiazacyclononadecino[11,12-b]quinoxaline-10-carboxamide hydrate.
The molecular formula is C38H46F4N6O9S (anhydrate) and the molecular weight for the drug substance is 838.87 g/mol (anhydrate). The strength of glecaprevir is based on anhydrous glecaprevir. Glecaprevir is a white to off-white crystalline powder with a solubility of less than 0.1 to 0.3 mg/mL across a pH range of 2–7 at 37°C and is practically insoluble in water, but is sparingly soluble in ethanol. Glecaprevir has the following molecular structure:
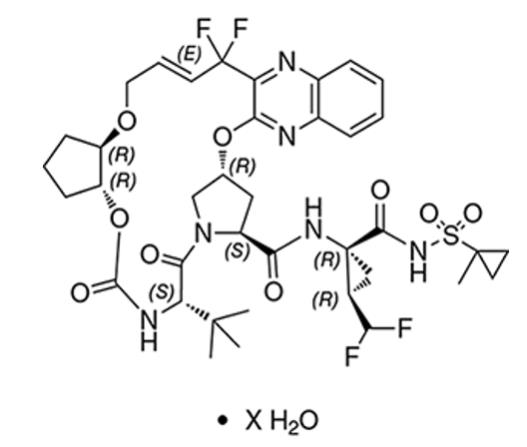
Pibrentasvir drug substance:
The chemical name of pibrentasvir is Methyl {(2S,3R)-1-[(2S)-2-{5-[(2R,5R)-1-{3,5-difluoro-4-[4-(4-fluorophenyl)piperidin-1-yl]phenyl}-5-(6-fluoro-2-{(2S)-1-[N-(methoxycarbonyl)-O-methyl-L-threonyl]pyrrolidin-2-yl}-1H-benzimidazol-5-yl)pyrrolidin-2-yl]-6-fluoro-1H-benzimidazol-2-yl}pyrrolidin-1-yl]-3-methoxy-1-oxobutan-2-yl}carbamate.
The molecular formula is C57H65F5N10O8 and the molecular weight for the drug substance is 1113.18 g/mol. Pibrentasvir is a white to off-white to light yellow crystalline powder with a solubility of less than 0.1 mg/mL across a pH range of 1–7 at 37°C and is practically insoluble in water, but is freely soluble in ethanol. Pibrentasvir has the following molecular structure:
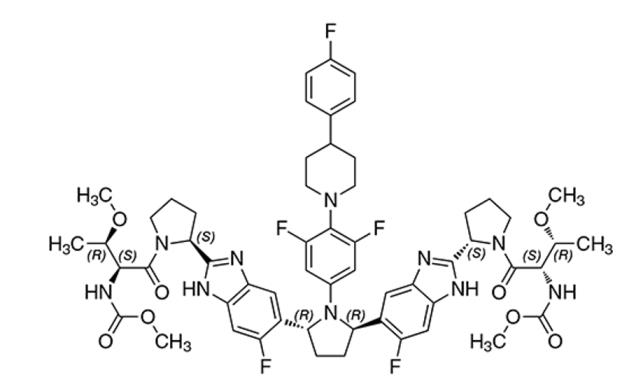
12. Clinical Pharmacology
12.1 Mechanism of Action
Mechanism of Action
MAVYRET is a fixed-dose combination of glecaprevir and pibrentasvir, which are direct-acting antiviral agents against the hepatitis C virus [see Microbiology ( 12.4 ) ].
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of doses up to glecaprevir 600 mg (2 times the recommended dosage) with doses up to pibrentasvir 240 mg (2 times the recommended dosage) on QTc interval was evaluated in an active-controlled (moxifloxacin 400 mg) thorough QT study. At 20-fold of glecaprevir and 5-fold of pibrentasvir therapeutic concentrations, the glecaprevir and pibrentasvir combination does not prolong the QTc interval to any clinically relevant extent.
12.3 Pharmacokinetics
The pharmacokinetic properties of the components of MAVYRET in healthy subjects are provided in Table 7. The steady-state pharmacokinetic parameters of glecaprevir and pibrentasvir in HCV-infected subjects without cirrhosis are provided in Table 8. For pellets relative to tablets in healthy adult subjects under non-fasting conditions, geometric mean ratios (GMRs) of glecaprevir and pibrentasvir Cmax were 0.664 and 1.137, AUCinf were 0.795 and 1.219, and C24 were 0.917 and 1.174. These differences were not considered clinically significant.
| Table 7. Pharmacokinetic Properties of the Components of MAVYRET in Healthy Subjects | ||
| Glecaprevir | Pibrentasvir | |
| Absorption | ||
| Tmax (h)a of tablets | 5.0 | 5.0 |
| Tmax (h)a of oral pellets | 3.0 | 5.0 |
| Effect of meal (relative to fasting)b on tablets | ↑ 83-163% | ↑ 40-53% |
| Effect of meal (relative to fasting)b on oral pellets | ↑ 131-167% | ↑ 56-114% |
| Distribution | ||
| % Bound to human plasma proteins | 97.5 | >99.9 |
| Blood-to-plasma ratio | 0.57 | 0.62 |
| Elimination | ||
| t1/2 (h) | 6 | 13 |
| Metabolism | secondary, | None |
| CYP3A | ||
| Major route of excretion | biliary-fecal | biliary-fecal |
| % of dose excreted in urinec | 0.7 | 0 |
| % of dose excreted in fecesc | 92.1 | 96.6 |
| a. Median Tmax following single doses of glecaprevir and pibrentasvir in healthy subjects. b. Mean systemic exposures with low/moderate to high fat meals. c. Single dose administration of radiolabeled glecaprevir or pibrentasvir in mass balance studies. | ||
| Table 8. Steady-State Pharmacokinetic Parameters of Glecaprevir and Pibrentasvir Following Administration of MAVYRET in Non-Cirrhotic HCV-Infected Subjects | ||
| Pharmacokinetic Parameter | Glecaprevirb | Pibrentasvirc |
| Cmax (ng/mL)a | 597 (114) | 110 (49) |
| AUC24,ss (ng•h/mL)a | 4800 (122) | 1430 (57) |
| Ctrough,ss (ng/mL)a | 13.0 (334) | 18.9 (92) |
| a Geometric mean (%CV) of individual-estimated Cmax, AUC24,ss and Ctrough,ss values b Relative to healthy subjects, glecaprevir Cmax was 51% lower, AUC24,ss was similar (10% difference), and Ctrough,ss was 157% higher in HCV-infected subjects without cirrhosis c Relative to healthy subjects, pibrentasvir Cmax was 63% lower, AUC24,ss was 34% lower, and Ctrough,ss was 37% lower in HCV-infected subjects without cirrhosis | ||
Specific Populations
Pediatric Patients
The pharmacokinetics of glecaprevir and pibrentasvir were determined in HCV-infected pediatric subjects 3 years of age and older receiving a daily dose of MAVYRET as described below in Table 9. GMRs of glecaprevir and pibrentasvir Cmax and AUC24 in HCV-infected pediatrics vs. adults ranged from 1.58-2.68 and 0.965-1.64, respectively. GMRs of glecaprevir Ctrough ranged from 0.292-0.954 and GMRs of pibrentasvir Ctrough ranged from 0.794-1.93. All pediatric glecaprevir and pibrentasvir PK parameter values fell within the range observed in adult subjects. These differences were not considered clinically significant. The pharmacokinetics of glecaprevir and pibrentasvir have not been established in children less than 3 years of age.
| Table 9. Pharmacokinetic Parameters of Glecaprevir (GLE) and Pibrentasvir (PIB) in HCV Infected Pediatric Subjects | |||||
| Age and Weight (kg) | N | Total Daily Dose of GLE/PIB (mg) | PK Parameter | Geometric Mean (%CV) | |
| GLE | PIB | ||||
| 12 to < 18 years, ≥ 45 kg | 14 | 300/120 | AUC24 (ng•h/mL) | 4790 (72) | 1380 (40) |
| Cmax (ng/mL) | 1040 (86) | 174 (36) | |||
| Ctrough (ng/mL) | 3.79 (82) | 15.0 (61) | |||
| 9 to < 12 years, 30 to < 45 kg | 13 | 250/100 | AUC24 (ng•h/mL) | 7870 (209) | 2200 (99) |
| Cmax (ng/mL) | 1370 (169) | 225 (72) | |||
| Ctrough (ng/mL) | 12.4 (856) | 36.5 (164) | |||
| 6 to < 9 years, 20 to < 30 kg | 13 | 200/80 | AUC24 (ng•h/mL) | 6860 (142) | 1640 (63) |
| Cmax (ng/mL) | 1600 (155) | 197 (52) | |||
| Ctrough (ng/mL) | 7.44 (383) | 19.4 (103) | |||
| 3 to < 6 years, 12 to < 20 kg | 12 | 150/60 | AUC24 (ng•h/mL) | 7520 (205) | 1790 (58) |
| Cmax (ng/mL) | 1530 (280) | 233 (48) | |||
| Ctrough (ng/mL) | 6.58 (318) | 17.9 (119) | |||
Subjects with Renal Impairment
Glecaprevir and pibrentasvir AUC were increased ≤ 56% in non-HCV infected subjects with mild, moderate, severe, or end-stage renal impairment (GFR estimated using Modification of Diet in Renal Disease) not on dialysis compared to subjects with normal renal function. Glecaprevir and pibrentasvir AUC were similar with and without dialysis (≤ 18% difference) in dialysis-dependent non-HCV infected subjects. In HCV-infected subjects, 86% higher glecaprevir and 54% higher pibrentasvir AUC were observed for subjects with end stage renal disease, with or without dialysis, compared to subjects with normal renal function.
Subjects with Hepatic Impairment
Following administration of MAVYRET in HCV infected subjects with compensated cirrhosis (Child-Pugh A), exposure of glecaprevir was approximately 2-fold and pibrentasvir exposure was similar to non-cirrhotic HCV infected subjects.
At the clinical dose, compared to non-HCV infected subjects with normal hepatic function, glecaprevir AUC was 100% higher in Child-Pugh B subjects, and increased 11-fold in Child-Pugh C subjects. Pibrentasvir AUC was 26% higher in Child-Pugh B subjects, and 114% higher in Child-Pugh C subjects.
Age/Gender/Race/Body Weight
No clinically significant differences in the pharmacokinetics of glecaprevir or pibrentasvir were observed based on age (12-88 years), sex, race/ethnicity or body weight (45 kg or greater). Patients under the age of 12 and weighing less than 45 kg are dosed based on body weight [see Dosage and Administration ( 2.4 ) ].
Drug Interaction Studies
Drug interaction studies were performed with glecaprevir/pibrentasvir and other drugs that are likely to be coadministered and with drugs commonly used as probes for pharmacokinetic interactions. Tables 10 and 11 summarize the pharmacokinetic effects when glecaprevir/pibrentasvir was coadministered with other drugs which showed potentially clinically relevant changes. Significant interactions are not expected when MAVYRET is coadministered with substrates of CYP3A, CYP1A2, CYP2C9, CYP2C19, CYP2D6, UGT1A1, or UGT1A4.
| Table 10. Drug Interactions: Changes in Pharmacokinetic Parameters of Glecaprevir (GLE) or Pibrentasvir (PIB) in the Presence of Coadministered Drug | |||||||
| Co-administeredDrug | Regimenof Co-administeredDrug (mg) | RegimenofGLE/PIB(mg) | N | DAA | Central Value Ratio(90% CI) | ||
| Cmax | AUC | Cmin | |||||
| Atazanavir + ritonavir | 300 + 100 once daily | 300/120 once dailya | 12 | GLE | ≥4.06 (3.15, 5.23) | ≥6.53 (5.24, 8.14) | ≥14.3 (9.85, 20.7) |
| PIB | ≥1.29 (1.15, 1.45) | ≥1.64 (1.48, 1.82) | ≥2.29 (1.95, 2.68) | ||||
| Carbamazepine | 200 twice daily | 300/120 single dose | 10 | GLE | 0.33 (0.27, 0.41) | 0.34 (0.28, 0.40) | -- |
| PIB | 0.50 (0.42, 0.59) | 0.49 (0.43, 0.55) | -- | ||||
| Cyclosporine | 100 single dose | 300/120 once daily | 12 | GLEb | 1.30 (0.95, 1.78) | 1.37 (1.13, 1.66) | 1.34 (1.12, 1.60) |
| PIB | ↔ | ↔ | 1.26 (1.15, 1.37) | ||||
| 400 single dose | 300/120 single dose | 11 | GLE | 4.51 (3.63, 6.05) | 5.08 (4.11, 6.29) | -- | |
| PIB | ↔ | 1.93 (1.78, 2.09) | -- | ||||
| Darunavir + ritonavir | 800 + 100 once daily | 300/120 once daily | 8 | GLE | 3.09 (2.26, 4.20) | 4.97 (3.62, 6.84) | 8.24 (4.40, 15.4) |
| PIB | ↔ | ↔ | 1.66 (1.25, 2.21) | ||||
| Elvitegravir/ cobicistat/ emtricitabine/ tenofovir alafenamide | 150/150/ 200/10 once daily | 300/120 once daily | 11 | GLE | 2.50 (2.08, 3.00) | 3.05 (2.55, 3.64) | 4.58 (3.15,6.65) |
| PIB | ↔ | 1.57 (1.39, 1.76) | 1.89 (1.63, 2.19) | ||||
| Omeprazole | 20 once daily | 300/120 single dose | 9 | GLE | 0.78 (0.60, 1.00) | 0.71 (0.58, 0.86) | -- |
| PIB | ↔ | ↔ | -- | ||||
| 40 once daily (1 hour before GLE/PIB) | 300/120 single dose | 12 | GLE | 0.36 (0.21, 0.59) | 0.49 (0.35, 0.68) | -- | |
| PIB | ↔ | ↔ | -- | ||||
| Rifampin | 600 (first dose) | 300/120 single dose | 12 | GLE | 6.52 (5.06, 8.41) | 8.55 (7.01, 10.4) | -- |
| PIB | ↔ | ↔ | -- | ||||
| 600 once daily | 300/120 single dosec | 12 | GLE | 0.14 (0.11, 0.19) | 0.12 (0.09, 0.15) | -- | |
| PIB | 0.17 (0.14, 0.20) | 0.13 (0.11, 0.15) | -- | ||||
| Lopinavir/ ritonavir | 400/100 twice daily | 300/120 once daily | 9 | GLE | 2.55 (1.84, 3.52) | 4.38 (3.02, 6.36) | 18.6 (10.4, 33.5) |
| PIB | 1.40 (1.17, 1.67) | 2.46 (2.07, 2.92) | 5.24 (4.18, 6.58) | ||||
| ↔ = No change (central value ratio 0.80 to 1.25) a. Effect of atazanavir and ritonavir on the first dose of glecaprevir and pibrentasvir is reported. b. HCV-infected transplant recipients who received cyclosporine dose of 100 mg or less per day had mean glecaprevir exposures 2.4-fold of those not receiving cyclosporine. c. Effect of rifampin on glecaprevir and pibrentasvir 24 hours after final rifampin dose. | |||||||
| Table 11. Drug Interactions: Pharmacokinetic Parameters for Coadministered Drug in the Presence of Combination of Glecaprevir/Pibrentasvir (GLE/PIB) | ||||||
| Co-administeredDrug | Regimenof Co-administeredDrug (mg) | Regimenof GLE/PIB(mg) | N | Central Value Ratio (90% CI) | ||
| Cmax | AUC | Cmin | ||||
| Abacavir | ABC/DTG/3TC 600/50/300 once daily | 300/120 once daily | 12 | ↔ | ↔ | 1.31 (1.05, 1.63) |
| Atorvastatin | 10 once daily | 400/120 once daily | 11 | 22.0 (16.4, 29.6) | 8.28 (6.06, 11.3) | -- |
| Caffeine | 100 single dose | 300/120 once daily | 12 | ↔ | 1.35 (1.23, 1.48) | -- |
| Dabigatran | Dabigatran etexilate 150 single dose | 300/120 once daily | 11 | 2.05 (1.72, 2.44) | 2.38 (2.11, 2.70) | -- |
| Darunavir | DRV + RTV 800 + 100 once daily | 300/120 once daily | 12 | 1.30 (1.21, 1.40) | 1.29 (1.18, 1.42) | ↔ |
| Ritonavir | 2.03 (1.78, 2.32) | 1.87 (1.74, 2.02) | ↔ | |||
| Dextro- methorphan | Dextromethorphan hydrobromide 30 single dose | 300/120 once daily | 12 | 0.70 (0.61, 0.81) | 0.75 (0.66, 0.85) | -- |
| Digoxin | 0.5 single dose | 400/120 once daily | 12 | 1.72 (1.45, 2.04) | 1.48 (1.40, 1.57) | -- |
| Ethinyl estradiol (EE) | EE/ Norgestimate 35 µg/250 µg once daily | 300/120 once daily | 11 | 1.31 (1.24, 1.38) | 1.28 (1.23, 1.32) | 1.38 (1.25, 1.52) |
| Norgestrel | 1.54 (1.34, 1.76) | 1.63 (1.50, 1.76) | 1.75 (1.62, 1.89) | |||
| Norgestromin | ↔ | 1.44 (1.34, 1.54) | 1.45 (1.33, 1.58) | |||
| Ethinyl estradiol | EE/ Levonorgestrel 20 µg/100 µg once daily | 300/120 once daily | 12 | 1.30 (1.18, 1.44) | 1.40 (1.33, 1.48) | 1.56 (1.41, 1.72) |
| Norgestrel | 1.37 (1.23, 1.52) | 1.68 (1.57, 1.80) | 1.77 (1.58, 1.98) | |||
| Elvitegravir | EVG/COBI/FTC/ TAF 150/ 150/200/10 once daily | 300/120 once daily | 12 | 1.36 (1.24, 1.49) | 1.47 (1.37, 1.57) | 1.71 (1.50, 1.95) |
| Tenofovir | ↔ | ↔ | ↔ | |||
| Felodipine | 2.5 single dose | 300/120 once daily | 11 | 1.31 (1.05, 1.62) | 1.31 (1.08, 1.58) | -- |
| Losartan | 50 single dose | 300/120 once daily | 12 | 2.51 (2.00, 3.15) | 1.56 (1.28, 1.89) | -- |
| Losartan carboxylic acid | 2.18 (1.88, 2.53) | ↔ | -- | |||
| Lovastatin | Lovastatin 10 once daily | 300/120 once daily | 12 | ↔ | 1.70 (1.40, 2.06) | -- |
| Lovastatin acid | 5.73 (4.65, 7.07) | 4.10 (3.45, 4.87) | -- | |||
| Midazolam | 1 single dose | 300/120 once daily | 12 | ↔ | 1.27 (1.11, 1.45) | -- |
| Omeprazole | 20 single dose | 300/120 once daily | 12 | 0.57 (0.43, 0.75) | 0.79 (0.70, 0.90) | -- |
| Pravastatin | 10 once daily | 400/120 once daily | 12 | 2.23 (1.87, 2.65) | 2.30 (1.91, 2.76) | -- |
| Raltegravir | 400 twice daily | 300/120 once daily | 12 | 1.34 (0.89, 1.98) | 1.47 (1.15, 1.87) | 2.64 (1.42, 4.91) |
| Rilpivirine | 25 once daily | 300/120 once daily | 12 | 2.05 (1.73, 2.43) | 1.84 (1.72, 1.98) | 1.77 (1.59, 1.96) |
| Rosuvastatin | 5 once daily | 400/120 once daily | 11 | 5.62 (4.80, 6.59) | 2.15 (1.88, 2.46) | -- |
| Simvastatin | Simvastatin 5 once daily | 300/120 once daily | 12 | 1.99 (1.60, 2.48) | 2.32 (1.93, 2.79) | -- |
| Simvastatin acid | 10.7 (7.88, 14.6) | 4.48 (3.11, 6.46) | -- | |||
| Sofosbuvir | Sofosbuvir 400 once daily | 400/120 once daily | 8 | 1.66 (1.23, 1.22) | 2.25 (1.86, 2.72) | -- |
| GS-331007 | ↔ | ↔ | 1.85 (1.67, 2.04) | |||
| Tacrolimus | 1 single dose | 300/120 once daily | 10 | 1.50 (1.24, 1.82) | 1.45 (1.24, 1.70) | -- |
| Tenofovir | EFV/FTC/TDF 300/200/300 once daily | 300/120 once daily | 12 | ↔ | 1.29 (1.23, 1.35) | 1.38 (1.31, 1.46) |
| Valsartan | 80 single dose | 300/120 once daily | 12 | 1.36 (1.17, 1.58) | 1.31 (1.16, 1.49) | -- |
| ↔ = No change (central value ratio 0.80 to 1.25) 3TC – lamivudine; ABC – abacavir; COBI – cobicistat; DRV – darunavir; DTG – dolutegravir; EFV – efavirenz; EVG – elvitegravir; FTC – emtricitabine; RTV – ritonavir; TAF – tenofovir alafenamide; TDF – tenofovir disoproxil fumarate | ||||||
12.4 Microbiology
Mechanism of Action
Glecaprevir
Glecaprevir is an inhibitor of the HCV NS3/4A protease, which is necessary for the proteolytic cleavage of the HCV encoded polyprotein (into mature forms of the NS3, NS4A, NS4B, NS5A, and NS5B proteins) and is essential for viral replication. In a biochemical assay, glecaprevir inhibited the proteolytic activity of recombinant NS3/4A enzymes from clinical isolates of HCV genotypes 1a, 1b, 2a, 2b, 3a, 4a, 5a, and 6a with IC50 values ranging from 3.5 to 11.3 nM.
Pibrentasvir
Pibrentasvir is an inhibitor of HCV NS5A, which is essential for viral RNA replication and virion assembly. The mechanism of action of pibrentasvir has been characterized based on cell culture antiviral activity and drug resistance mapping studies.
Antiviral Activity
In HCV replicon assays, glecaprevir had median EC50 values of 0.08-4.6 nM against laboratory and clinical isolates from subtypes 1a, 1b, 2a, 2b, 3a, 3b, 4a, 4d, 5a, and 6a. Pibrentasvir had median EC50 values of 0.5-15.6 pM against laboratory and clinical isolates from subtypes 1a, 1b, 2a, 2b, 3a, 3b, 4a, 4b, 4d, 5a, 6a, 6e and 6p.
Combination Antiviral Activity
Evaluation of combination of glecaprevir and pibrentasvir showed no antagonism in antiviral activity in HCV genotype 1 replicon cell culture assays.
Resistance
In Cell Culture
Selection of HCV genotype 1a, 1b, 2a, 3a, 4a or 6a replicons for reduced susceptibility to glecaprevir resulted in the emergence of amino acid substitutions most commonly at NS3 positions A156 or D/Q168. Individual substitutions at NS3 amino acid position A156 introduced into HCV replicons by site-directed mutagenesis generally caused the greatest reductions (>100-fold) in susceptibility to glecaprevir. Individual substitutions at NS3 position D/Q168 had varying effects on glecaprevir susceptibility depending on HCV genotype/subtype and specific amino acid change, with the greatest reductions (>30-fold) observed in genotypes 1a (D168F/Y), 3a (Q168R) and 6a (D168A/G/H/V/Y). Combinations of NS3 Y56H plus D/Q168 substitutions resulted in greater reductions in glecaprevir susceptibility. An NS3 Q80R substitution in genotype 3a caused a 21-fold reduction in glecaprevir susceptibility, while Q80 substitutions in genotypes 1a and 1b (including genotype 1a Q80K) did not reduce glecaprevir susceptibility. Individual amino acid substitutions associated with resistance to other HCV protease inhibitors at positions 36, 43, 54, 55, 56, 155, 166, or 170 in NS3 generally did not reduce susceptibility to glecaprevir.
Selection of HCV genotype 1a, 2a or 3a replicons for reduced susceptibility to pibrentasvir resulted in the emergence of amino acid substitutions at known NS5A inhibitor resistance-associated positions, including Q30D/deletion, Y93D/H/N or H58D +Y93H in genotype 1a replicons, F28S + M31I or P29S + K30G in genotype 2a replicons, and Y93H in genotype 3a replicons. The majority of individual amino acid substitutions associated with resistance to other HCV NS5A inhibitors at positions 24, 28, 30, 31, 58, 92, or 93 in NS5A did not reduce susceptibility to pibrentasvir. Individual NS5A amino acid substitutions that reduced susceptibility to pibrentasvir include M28G or Q30D in a genotype 1a replicon (244- and 94-fold, respectively), and P32-deletion in a genotype 1b replicon (1,036-fold). Some combinations of two or more NS5A inhibitor resistance-associated amino acid substitutions may result in greater reductions in pibrentasvir susceptibility. In a genotype 3b replicon, the presence of naturally occurring polymorphisms K30 and M31 in NS5A reduced susceptibility to pibrentasvir by 24-fold relative to the activity of pibrentasvir in a genotype 3a replicon. Introduction of an NS5A Y93H substitution into a genotype 3b replicon further reduced susceptibility to pibrentasvir by 6336-fold.
In Clinical Studies
Studies in Treatment-Naïve and (peg)Interferon, Ribavirin and/or Sofosbuvir Treatment-Experienced Subjects with or without Cirrhosis
In pooled analyses of NS3/4A PI- and NS5A inhibitor-naïve subjects who received MAVYRET for 8, 12, or 16 weeks in the registrational Phase 2 and 3 clinical studies (including EXPEDITION-2 and MAGELLAN-2), treatment-emergent resistance analyses were conducted for 24 subjects who experienced virologic failure (2 with genotype 1, 2 with genotype 2, 20 with genotype 3 infection). No subjects with HCV genotype 4, 5 or 6 infection experienced virologic failure.
Among the two genotype 1-infected subjects who experienced virologic failure, both subjects had a subtype 1a infection. One subject had treatment-emergent substitutions A156V in NS3, and Q30R, L31M and H58D in NS5A (Q30R and L31M were also detected at a low frequency at baseline). One subject had treatment-emergent Q30R and H58D (while Y93N was present at baseline and post-treatment) in NS5A.
Among the two genotype 2-infected subjects who experienced virologic failure, both subjects had a subtype 2a infection, and no treatment-emergent substitutions were observed in NS3 or NS5A.
Among the 20 genotype 3-infected subjects who experienced virologic failure, treatment-emergent NS3 substitutions Y56H/N, Q80K/R, A156G, or Q168L/R were observed in 13 subjects. A166S or Q168R were present at baseline and post-treatment in 5 subjects. Treatment-emergent NS5A substitutions S24F, M28G/K, A30G/K, L31F, P58T, or Y93H were observed in 17 subjects, and 14 subjects had A30K (n=9) or Y93H (n=6) at baseline and post-treatment.
Studies in Subjects with or without Cirrhosis Who Were Treatment-Experienced to NS3/4A Protease and/or NS5A Inhibitors
Treatment-emergent resistance analyses were conducted for 11 HCV genotype 1-infected subjects (10 genotype 1a, 1 genotype 1b) with prior NS3/4A PI or NS5A inhibitor treatment experience who experienced virologic failure with MAVYRET with or without ribavirin in the MAGELLAN-1 study. Treatment-emergent NS3 substitutions V36A/M, Y56H, R155K/T, A156G/T/V, or D168A/T were observed in 73% (8/11) of subjects. Nine of 10 subjects (90%, not including one subject missing NS5A data at failure) had treatment-emergent NS5A substitutions M28A/G (or L28M for genotype 1b), P29Q/R, Q30K/R, H58D or Y93H/N. All 11 subjects also had NS5A inhibitor resistance-associated substitutions detected at baseline, and 7/11 had NS3 PI resistance-associated substitutions detected at baseline (see Cross-Resistance for the effect of baseline resistance-associated substitutions on treatment response for NS3/4A PI or NS5A inhibitor treatment-experienced patients).
Effect of Baseline HCV Amino Acid Polymorphisms on Treatment Response (NS3/4A PI- and NS5A Inhibitor-Naïve Subjects)
A pooled analysis of NS3/4A PI- and NS5A inhibitor-naïve subjects who received MAVYRET in the Phase 2 and Phase 3 clinical studies was conducted to identify the HCV subtypes represented and explore the association between baseline amino acid polymorphisms and treatment outcome. Baseline polymorphisms relative to a subtype-specific reference sequence at resistance-associated amino acid positions 155, 156, and 168 in NS3, and 24, 28, 30, 31, 58, 92, and 93 in NS5A were evaluated at a 15% detection threshold by next-generation sequencing. Among subjects who received MAVYRET for 8-, 12-, or 16 weeks, baseline polymorphisms in NS3 were detected in 1% (9/845), 1% (3/398), 2% (10/613), 1% (2/164), 42% (13/31), and 3% (1/34) of subjects with HCV genotype 1, 2, 3, 4, 5, and 6 infection, respectively. No baseline polymorphisms were detected at NS3 amino acid position 156 across all genotypes. Baseline polymorphisms in NS5A were detected in 27% (225/841), 80% (331/415), 22% (136/615), 50% (80/161), 13% (4/31), and 54% (20/37) of subjects with HCV genotype 1, 2, 3, 4, 5, and 6 infection, respectively.
Genotype 1, 2, 4, 5, and 6: Baseline HCV polymorphisms in genotypes 1, 2, 4, 5 and 6 had no impact on treatment outcome.
Genotype 3: In registrational trials, HCV subtype 3a was the predominant genotype 3 subtype overall and was detected in >99% of U.S. genotype 3-infected subjects. Among treatment-naïve, genotype 3a-infected subjects without cirrhosis who received MAVYRET for 8 weeks, an NS5A A30K polymorphism was detected in 10% (18/179) of subjects, of whom 78% (14/18) achieved SVR12. Limited data are available to characterize the impact of the A30K polymorphism in genotype 3a-infected subjects with cirrhosis (n=3 who received MAVYRET for 8 weeks, all achieved SVR12) or prior treatment experience (n=1 who received MAVYRET for 16 weeks, relapse). In the pooled Phase 2 and Phase 3 trials, including post-registrational trials EXPEDITION-8, VOYAGE-1 and VOYAGE-2, 94% (15/16) of genotype 3a-infected subjects with Y93H in NS5A at baseline who received the recommended MAVYRET regimens achieved SVR12. In MAGELLAN-2 (post-transplant subjects), SVR12 was achieved in 2 of 3 (67%) genotype 3-infected subjects with the NS5A Y93H baseline polymorphism. In the VOYAGE-1 and VOYAGE-2 trials conducted in China, Singapore, and South Korea, 50% (20/40) of genotype 3-infected subjects had subtype 3b, of whom 14 (70%) achieved SVR12 with MAVYRET durations of 8, 12 or 16 weeks [see Clinical Studies ( 14.4 )]. The naturally occurring NS5A K30 and M31 polymorphisms were detected in 95% (19/20) and 100% (20/20) of genotype 3b-infected subjects.
Cross-resistance
Based on resistance patterns observed in cell culture replicon studies and HCV-infected subjects, cross-resistance is possible between glecaprevir and other HCV NS3/4A PIs, and between pibrentasvir and other HCV NS5A inhibitors. Cross-resistance is not expected between MAVYRET and sofosbuvir, (peg)interferon or ribavirin.
In the MAGELLAN-1 study, HCV genotype 1-infected subjects who had failed prior treatment with NS3/4A protease and/or NS5A inhibitors were treated with MAVYRET for 12 or 16 weeks. Baseline sequences were analyzed by next generation sequencing at a 15% detection threshold.
Among 23 NS3/4A PI-experienced/NS5A inhibitor-naïve subjects who received MAVYRET for 12 weeks in MAGELLAN-1 (excluding 2 non-virologic failure subjects), 2 subjects each had baseline NS3 R155K or D168E/V substitutions; all 23 subjects achieved SVR12.
Among NS5A inhibitor-experienced/PI-naïve subjects who received MAVYRET for 16 weeks, baseline NS5A resistance-associated substitutions [R30Q (n=1), Y93H/N (n=5), M28A+Q30R (n=1), Q30H+Y93H (n=1), Q30R+L31M (n=2), L31M+H58P (n=1)], were detected in 73% (11/15) of subjects with available data, of whom 91% (10/11) achieved SVR12. The non-SVR12 subject experienced on-treatment virologic failure and had a genotype 1a infection with baseline NS5A Q30R and L31M substitutions.
Persistence of Resistance-Associated Substitutions
Data on the persistence of glecaprevir and pibrentasvir resistance-associated substitutions are not available. NS5A resistance-associated substitutions observed in patients treated with other NS5A inhibitors have been found to persist for longer than 1 year. In patients treated with other NS3/4A PI, viral populations with NS3 resistance-associated substitutions have been found to decline in some patients through post-treatment weeks 24 and 48. The long-term clinical impact of the emergence or persistence of virus containing glecaprevir or pibrentasvir resistance-associated substitutions is unknown.
13. Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
Glecaprevir and pibrentasvir were not genotoxic in a battery of in vitro or in vivo assays, including bacterial mutagenicity, chromosome aberration using human peripheral blood lymphocytes and in vivo rodent micronucleus assays.
Carcinogenicity studies with glecaprevir and pibrentasvir have not been conducted.
Impairment of Fertility
No effects on mating, female or male fertility, or early embryonic development were observed in rodents at up to the highest dose tested. Systemic exposures (AUC) to glecaprevir and pibrentasvir were approximately 63 and 102 times higher, respectively, than the exposure in humans at the recommended dose.
14. Clinical Studies
14.1 Description of Clinical Trials
Table 12 summarizes the clinical trials conducted to support the effectiveness of MAVYRET in subjects with HCV genotype 1, 2, 3, 4, 5 or 6 infection and compensated liver disease (including Child-Pugh A cirrhosis) according to treatment history and cirrhosis status.
| Table 12. Clinical Trials Conducted with MAVYRET in Subjects with HCV Genotype 1, 2, 3, 4, 5 or 6 Infection and Compensated Liver Disease | ||
| Genotype(GT) | Clinical Trial(NCT Number) | Treatment Duration* |
| TN and PRS-TE Subjects without Cirrhosis | ||
| GT1** | ENDURANCE-1 (NCT02604017) | MAVYRET for 8 (n=351) or 12 weeks (n=352) |
| GT2 | SURVEYOR-2 (NCT02243293) | MAVYRET for 8 weeks (n=197) |
| GT3 | ENDURANCE-3 (NCT02640157) | MAVYRET for 8 (n=157) or 12 weeks (n=233) sofosbuvir + daclatasvir for 12 weeks (n=115) |
| SURVEYOR-2 | MAVYRET for 16 (PRS-TE only) weeks (n=22) | |
| GT4, 5, 6 | ENDURANCE-5,6 (NCT02966795) | MAVYRET for 8 weeks (GT5 n=20; GT6 n=55) |
| SURVEYOR-2 | MAVYRET for 8 weeks (GT4 n=46; GT5 n=2; GT6 n=10) | |
| GT1, 2, 3, 6 | VOYAGE-1 (NCT03222583) | MAVYRET for 8 (n=356) or 16 weeks (n=6; GT3 PRS-TE only) |
| TN and PRS-TE Subjects with Compensated Cirrhosis | ||
| GT1, 2, 4, 5, 6 | EXPEDITION-1 (NCT02642432) | MAVYRET for 12 weeks (n=146) |
| GT1, 2, 3, 4, 5, 6 | EXPEDITION-8 (NCT03089944) | MAVYRET for 8 weeks (n=343) (TN only) |
| GT3 | SURVEYOR-2 | MAVYRET for 16 weeks (PRS-TE only) (n=47) |
| GT5, 6 | ENDURANCE-5,6 | MAVYRET for 12 weeks (GT 5 n=3; GT 6 n=6) |
| GT1, 2, 3, 4, 6 | VOYAGE-2 (NCT03235349) | MAVYRET for 12 (n=157) or 16 weeks (n=3; GT3 PRS-TE only) |
| Subjects with CKD Stage 4 and 5 without Cirrhosis or with Compensated Cirrhosis | ||
| GT1-6 | EXPEDITION-4 (NCT02651194) | MAVYRET for 12 weeks (n=104) |
| NS5A Inhibitor or PI-Experienced Subjects without Cirrhosis or with Compensated Cirrhosis | ||
| GT1 | MAGELLAN-1 (NCT02446717) | MAVYRET for 12 (n=25) or 16 weeks (n=17) |
| HCV/HIV-1 Co-Infected Subjects without Cirrhosis or with Compensated Cirrhosis | ||
| GT1, 2, 3, 4, 6 | EXPEDITION-2 (NCT02738138) | MAVYRET for 8 (n=137) or 12 weeks (n=16) |
| Liver or Kidney Transplant Recipients without Cirrhosis | ||
| GT1, 2, 3, 4, 6 | MAGELLAN-2 (NCT02692703) | MAVYRET for 12 weeks (n=100) |
| Adolescent Subjects (12 to less than 18 years) | ||
| GT1, 2, 3, 4** | DORA (Part 1) (NCT03067129) | MAVYRET for 8 (n=44) or 16 weeks (n=3) |
| Pediatric Subjects (3 to less than 12 years) | ||
| GT1, 2, 3, 4** | DORA (Part 2) (NCT03067129) | MAVYRET for 8 (n=78), 12 (n=1), or 16 weeks (n=1) |
| TN=treatment naïve; PI=protease inhibitor; CKD=chronic kidney disease PRS-TE= defined as prior treatment experience with regimens containing (peg)interferon, ribavirin, and/or sofosbuvir, but no prior treatment experience with an HCV NS3/4A PI or NS5A inhibitor. * Treatment durations for some trial arms shown in this table do not reflect recommended dosing for the respective genotypes, prior treatment history, and/or cirrhosis status. For recommended dosing in adults and pediatric patients 3 years and older [see Dosage and Administration ( 2.2 , 2.3 , 2.4 ) ]. ** ENDURANCE-1 included 33 subjects co-infected with HIV-1. DORA included 3 subjects co-infected with HIV-1. | ||
Serum HCV RNA values were measured during the clinical trials using the Roche COBAS AmpliPrep/COBAS TaqMan HCV test (version 2.0) with a lower limit of quantification (LLOQ) of 15 IU/mL (except for SURVEYOR-2 which used the Roche COBAS TaqMan real-time reverse transcriptase-PCR (RT-PCR) assay v. 2.0 with an LLOQ of 25 IU/mL). The primary endpoint across all clinical trials was sustained virologic response (SVR12), defined as HCV RNA less than LLOQ at 12 weeks after the end of treatment. Relapse was defined as HCV RNA ≥ LLOQ after end-of-treatment response among subjects who completed treatment. Subjects with missing HCV RNA data, such as those who discontinued due to an adverse event, subject withdrawal or were lost to follow-up, were counted as SVR12 failures.
Demographics and Baseline Characteristics of Clinical Trials in Adults Who Were Treatment-Naïve or Treatment-Experienced to (peg)Interferon, Ribavirin and/or Sofosbuvir (PRS) without Cirrhosis or with Compensated Cirrhosis (Child-Pugh A)
Of the 2,152 subjects without cirrhosis or with compensated cirrhosis who were treatment-naïve or treatment-experienced to combinations of (peg)interferon, ribavirin and/or sofosbuvir (PRS), treated in the registrational studies excluding EXPEDITION-4 and MAGELLAN-1, the median age was 54 years (range: 19 to 88); 73% were treatment-naïve, 27% were PRS treatment-experienced; 39% were HCV genotype 1; 21% were HCV genotype 2; 29% were HCV genotype 3; 7% were HCV genotype 4; 4% were HCV genotype 5, or 6; 13% were ≥65 years; 54% were male; 5% were Black; 12% had cirrhosis; 20% had a body mass index of at least 30 kg per m2; and median baseline HCV RNA level was 6.2 log10 IU/mL.
14.2 Treatment-Naïve or PRS Treatment-Experienced Adults with HCV Genotype 1, 2, 4, 5, or 6 Infection without Cirrhosis
The efficacy of MAVYRET in subjects who were treatment-naïve or treatment-experienced to combinations of (peg)interferon, ribavirin and/or sofosbuvir (PRS) with genotype 1, 2, 4, 5, or 6 chronic HCV infection without cirrhosis was studied in three trials using an 8-week duration: ENDURANCE-1, ENDURANCE-5,6, and SURVEYOR-2 [(Part 2 and Part 4)].
ENDURANCE-1 was a randomized (1:1), open-label, multi-national trial comparing the efficacy of 8 weeks of treatment with MAVYRET versus 12 weeks of treatment in subjects without cirrhosis with genotype 1 infection with or without HIV-1 co-infection (n=33 co-infected). Table 13 presents SVR12 in MAVYRET-treated genotype 1-infected subjects for the 8-week treatment arm. Due to numerically similar efficacy, MAVYRET is recommended for 8 weeks for treatment-naïve and PRS treatment-experienced genotype 1 subjects without cirrhosis, rather than 12 weeks [see Dosage and Administration ( 2.2 )].
| Table 13. ENDURANCE-1: Efficacy in Treatment-Naïve and PRS Treatment-Experienced Adults with HCV Genotype 1 Infection without Cirrhosis | |
| MAVYRET 8 Weeks | |
| GT1N=351 | |
| SVR12 | 99% (348/351) |
| Outcome for Subjects without SVR12 | |
| On-treatment VF | <1% (1/351) |
| Relapse | 0/349 |
| Other* | <1% (2/351) |
| VF= virologic failure * Includes subjects who discontinued due to adverse event, lost to follow-up, or subject withdrawal. | |
The SVR12 data from the open-label trials SURVEYOR-2 (Parts 2 and 4) and ENDURANCE-5,6 are pooled by genotype, where appropriate, in Table 1 4 for ease of display.
| Table 14. SURVEYOR-2 (Part 2 and Part 4) and ENDURANCE-5, 6: Efficacy in Treatment-Naïve and PRS Treatment-Experienced Adults with HCV Genotypes 2, 4, 5 or 6 Infection without Cirrhosis | ||||
| MAVYRET 8 Weeks | ||||
| GT2N=197 | GT4N=46 | GT5N=22 | GT6N=65 | |
| SVR 12 | 98% (193/197) | 93% (43/46) | 95% (21/22) | 100% (65/65) |
| Outcome for Subjects without SVR12 | ||||
| On Treatment VF | 0/197 | 0/46 | 0/22 | 0/65 |
| Relapse | 1% (2/195) | 0/45 | 5% (1/22) | 0/65 |
| Other* | 1% (2/197) | 7% (3/46) | 0/22 | 0/65 |
| GT=genotype; VF= virologic failure * Includes subjects who discontinued due to adverse event, lost to follow-up, or subject withdrawal. | ||||
14.3 Treatment-Naïve Adults with HCV Genotype 1-6 Infection with Compensated Cirrhosis or PRS Treatment-Experienced Adults with HCV Genotype 1, 2, 4, 5, or 6 Infection with Compensated Cirrhosis
The efficacy of MAVYRET in treatment-naïve subjects with genotype 1, 2, 3, 4, 5 or 6 chronic HCV infection and compensated cirrhosis (Child-Pugh A) was studied in EXPEDITION-8, a single-arm, open-label trial in 343 subjects who received MAVYRET for 8 weeks.
| Table 15. EXPEDITION-8: Efficacy in Treatment-Naïve Adults with HCV Genotype 1, 2, 3, 4, 5 or 6 Infection with Compensated Cirrhosis | |||||||
| MAVYRET 8 Weeks(N=343) | |||||||
| Total(all GTs)(N=343) | GT1(N=231) | GT2(N=26) | GT3(N=63) | GT4(N=13) | GT5(N=1) | GT6(N=9) | |
| SVR12 | 98% (335/343) | 98% (226/231) | 100% (26/26) | 95% (60/63) | 100% (13/13) | 100% (1/1) | 100% (9/9) |
| Outcome for Subjects without SVR12 | |||||||
| On-treatment VF | 0/343 | 0/231 | 0/26 | 0/63 | 0/13 | 0/1 | 0/9 |
| Relapse | <1% (1/336) | 0/225 | 0/26 | 2% (1/62) | 0/13 | 0/1 | 0/9 |
| Other* | 2% (7/343) | 2% (5/231) | 0/26 | 3% (2/63) | 0/13 | 0/1 | 0/9 |
| GT = genotype; VF = virologic failure * Includes subjects who discontinued due to lost to follow-up or subject withdrawal. | |||||||
The efficacy of MAVYRET in treatment-naive or PRS treatment-experienced subjects with genotype 1, 2, 4, 5 or 6 chronic HCV infection with compensated cirrhosis (Child-Pugh A) was studied in EXPEDITION-1 a single-arm, open-label trial, which included 146 subjects (TN N=110, TE-PRS N=36) treated with MAVYRET for 12 weeks, and in ENDURANCE-5, 6, an open-label trial in 84 subjects (TN N= 76, TE-PRS N=8) with genotype 5 or 6 chronic HCV infection, 9 of whom had compensated cirrhosis (GT5 N=3, GT6 N=6) and received MAVYRET for 12 weeks.
| Table 16. EXPEDITION-1 and ENDURANCE-5, 6: Efficacy in Treatment-Naïve and PRS Treatment-Experienced Adults with HCV Genotype 1, 2, 4, 5 or 6 Infection with Compensated Cirrhosis | ||||||
| MAVYRET 12 Weeks | ||||||
| Total(all GTs)(N=155) | GT1(N=90) | GT2(N=31) | GT4(N=16) | GT5(N=5) | GT6(N=13) | |
| SVR12 | 99% (153/155) | 99% (89/90) | 100% (31/31) | 100% (16/16) | 100% (5/5) | 92% (12/13) |
| Outcome for Subjects without SVR12 | ||||||
| On-treatment VF | <1% (1/155) | 0/90 | 0/31 | 0/16 | 0/5 | 8% (1/13) |
| Relapse | <1% (1/152) | 1% (1/88) | 0/31 | 0/16 | 0/5 | 0/12 |
| GT = genotype; VF = virologic failure | ||||||
14.4 Treatment-Naïve or PRS Treatment-Experienced Adults with HCV Genotype 3 Infection without Cirrhosis or with Compensated Cirrhosis
The efficacy of MAVYRET in subjects who were treatment-naïve or treatment-experienced to combinations of (peg)interferon, ribavirin and/or sofosbuvir (PRS) with genotype 3 chronic HCV infection without cirrhosis or with compensated cirrhosis was studied in ENDURANCE-3, EXPEDITION-8 and in SURVEYOR-2 Part 3. Subjects with genotype 3 HCV infection were also included in two Asian regional studies, VOYAGE-1 and VOYAGE-2.
ENDURANCE-3 was a partially-randomized, open-label, active-controlled trial in treatment-naïve subjects without cirrhosis. Subjects were randomized (2:1) to either MAVYRET for 12 weeks or to the combination of sofosbuvir and daclatasvir for 12 weeks; subsequently the trial included a third non-randomized arm with MAVYRET for 8 weeks. The SVR12 data are summarized in Table 1 7. Due to numerically similar efficacy, MAVYRET is recommended for 8 weeks for treatment-naïve genotype 3 subjects without cirrhosis, rather than 12 weeks [see Dosage and Administration ( 2.2 )].
| Table 17. ENDURANCE-3: Efficacy in Treatment-Naïve, HCV Genotype 3-Infected Adults without Cirrhosis | |||
| MAVYRET18 Weeks(N=157) | MAVYRET12 Weeks*(N=233) | DCV+SOF12 Weeks(N=115) | |
| SVR12 | 95% (149/157) | 95% (222/233)* | 97% (111/115) |
| Outcome for Subjects without SVR12 | |||
| On-treatment VF | 1% (1/157) | <1% (1/233) | 0/115 |
| Relapse | 3% (5/150) | 1% (3/222) | 1% (1/114) |
| Other2 | 1% (2/157) | 3% (7/233) | 3% (3/115) |
| VF=virologic failure 1 MAVYRET 8 weeks was a non-randomized treatment arm. 2 Includes subjects who discontinued due to adverse event, lost to follow-up, or subject withdrawal. * Data for MAVYRET 12-week treatment is displayed to reflect the original randomized study design. The treatment difference (95% confidence interval) was -1.2% (-5.6, 3.1) between the randomized arms of MAVYRET 12 weeks and DCV + SOF 12 weeks. | |||
The efficacy of MAVYRET in subjects who were treatment-naïve with genotype 3 chronic HCV infection and compensated cirrhosis was studied in EXPEDITION-8. The SVR12 rate of the treatment-naïve subjects with genotype 3 and compensated cirrhosis was 95% (60/63) and one subject experienced virologic relapse [see Clinical Studies ( 14.3 )].
SURVEYOR-2 Part 3 was an open-label trial randomizing PRS treatment-experienced subjects with genotype 3 infection without cirrhosis to 16-weeks of treatment. In addition, the trial evaluated the efficacy of MAVYRET in PRS treatment-experienced genotype 3-infected subjects with compensated cirrhosis for a 16-week duration. Among PRS treatment-experienced subjects treated with MAVYRET for 16 weeks, 49% (34/69) had failed a previous regimen containing sofosbuvir.
| Table 18. SURVEYOR-2 Part 3: Efficacy in PRS Treatment-Experienced, HCV Genotype 3-Infected Adults without Cirrhosis or with Compensated Cirrhosis | |
| PRS Treatment-Experiencedwithout Cirrhosis or With Compensated Cirrhosis | |
| MAVYRET 16 Weeks(N=69) | |
| SVR12 | 96% (66/69) |
| Outcome for Subjects without SVR12 | |
| On-treatment VF | 1% (1/69) |
| Relapse | 3% (2/68) |
| Other* | 0/69 |
| SVR12 by Cirrhosis Status | |
| Without Cirrhosis | 95% (21/22) |
| With Compensated Cirrhosis | 96% (45/47) |
| VF=virologic failure * Includes subjects who discontinued due to adverse event, lost to follow-up, or subject withdrawal. | |
Subjects with Genotype 3b Infection in VOYAGE-1 and VOYAGE-2
The efficacy of MAVYRET in subjects with HCV subtype 3b infection was evaluated in the VOYAGE-1 and VOYAGE-2 trials. Genotype 3b is a subtype that is uncommon in the U.S. (<1% of HCV GT3 infections) but has been reported in China, India and other countries in South and Southeast Asia. VOYAGE-1 and VOYAGE-2 were conducted in China, Singapore, and South Korea in HCV genotype 1, 2, 3, 4 or 6 infected subjects without cirrhosis (VOYAGE-1) or with compensated cirrhosis (VOYAGE-2) who were treatment-naive or PRS-treatment-experienced. All subjects without cirrhosis or with compensated cirrhosis received 8 or 12 weeks of MAVYRET, respectively, except genotype 3 PRS-treatment-experienced subjects who received 16 weeks of MAVYRET.
Across both trials, subjects with HCV genotype 3b infection had a numerically lower SVR12 rate of 70% (14/20) [58% (7/12) for non-cirrhotic subjects and 88% (7/8) for subjects with compensated cirrhosis] compared to subjects infected with genotype 3a or other HCV genotypes. All six genotype 3b subjects without SVR12 experienced virologic failure (2 on-treatment virologic failure, 4 relapse). SVR12 results in subjects with genotype 3a or other HCV genotypes were comparable with other trials.
14.5 Treatment-Naïve and PRS Treatment-Experienced Adults with CKD Stage 4 and 5 and Chronic HCV Infection without Cirrhosis or with Compensated Cirrhosis
EXPEDITION-4 was an open-label, single-arm, multicenter trial to evaluate safety and efficacy in subjects with severe renal impairment (CKD Stages 4 and 5) with compensated liver disease (with and without Child-Pugh A cirrhosis). There were 104 subjects enrolled, 82% were on hemodialysis, and 53%, 15%, 11%, 19%, 1% and 1% were infected with HCV genotypes 1, 2, 3, 4, 5 and 6, respectively. Overall, 19% of subjects had compensated cirrhosis and 81% of subjects were non-cirrhotic; 58% and 42% of subjects were treatment-naïve and PRS treatment-experienced, respectively. The overall SVR12 rate was 98% and no subjects experienced virologic failure. The presence of renal impairment did not affect efficacy; no dose-adjustments were required during the trial.
14.6 Adults Who are NS5A Inhibitor or NS3/4A-Protease Inhibitor (PI)-Experienced, without Cirrhosis or with Compensated Cirrhosis
MAGELLAN-1 was a randomized, multipart, open-label trial in 141 genotype 1- or 4-infected subjects who failed a previous regimen containing an NS5A inhibitor and/or NS3/4A PI. Part 1 (n=50) was a randomized trial exploring 12 weeks of glecaprevir 200 mg and pibrentasvir 80 mg, glecaprevir 300 mg and pibrentasvir 120 mg, with and without ribavirin (only data from glecaprevir 300 mg plus pibrentasvir 120 mg without ribavirin are included in these analyses). Part 2 (n=91) randomized genotype 1- or 4-infected subjects without cirrhosis or with compensated cirrhosis to 12- or 16-weeks of treatment with MAVYRET.
Of the 42 genotype 1-infected subjects treated in Parts 1 and 2, who were either NS5A inhibitor-experienced only (and treated for 16 weeks), or NS3/4A PI-experienced only (and treated for 12 weeks), the median age was 58 years (range: 34 to 70); 40% of the subjects were NS5A-treatment experienced only and 60% were PI experienced only; 24% had cirrhosis; 19% were ≥65 years, 69% were male; 26% were Black; 43% had a body mass index ≥ 30 kg/m2; 67% had baseline HCV RNA levels of at least 1,000,000 IU per mL; 79% had subtype 1a infection, 17% had subtype 1b infection and 5% had non-1a/1b infection.
Due to higher rates of virologic failure and treatment-emergent drug resistance, the data do not support labeling for treatment of HCV genotype 1-infected patients who are both NS3/4A PI and NS5A inhibitor-experienced.
| Table 19. MAGELLAN-1: Efficacy in HCV Genotype 1-Infected Adults Who Are NS3/4A PI-Experienced or NS5A Inhibitor-Experienced, without Cirrhosis or with Compensated Cirrhosis | ||
| PI-Experienced1(NS5A Inhibitor-naïve) | NS5A Inhibitor-Experienced2(PI-naïve) | |
| MAVYRET 12 Weeks (N=25) | MAVYRET 16 Weeks (N=17) | |
| SVR12 | 92% (23/25) | 94% (16/17) |
| Outcome for Subjects without SVR | ||
| On-treatment Virologic Failure | 0/25 | 6% (1/17) |
| Relapse | 0/25 | 0/16 |
| Other3 | 8% (2/25) | 0/17 |
| PI= protease inhibitor 1 Includes subjects who were treated with a regimen containing an NS3/4A PI (simeprevir with sofosbuvir, or simeprevir, boceprevir, or telaprevir with (peg)interferon and ribavirin) and without prior treatment with an NS5A inhibitor. 2 Includes subjects who were treated with a regimen containing an NS5A inhibitor (ledipasvir with sofosbuvir or daclatasvir with (peg)interferon and ribavirin) and without prior treatment with an NS3/4A PI. 3 Includes subjects who discontinued due to adverse event, lost to follow-up, or subject withdrawal. | ||
14.7 Treatment-Naïve or PRS Treatment-Experienced Adults with HCV/HIV-1 Coinfection without Cirrhosis or with Compensated Cirrhosis
EXPEDITION-2 was an open-label study in 153 HCV/HIV-1-coinfected subjects. Subjects without cirrhosis received MAVYRET for 8 weeks and subjects with compensated cirrhosis received MAVYRET for 12 weeks. The study included subjects who were HCV treatment-naïve or treatment-experienced to combinations of (peg)interferon, ribavirin, and/or sofosbuvir, with the exception of genotype 3-infected subjects who were all treatment naïve.
Of the 153 subjects treated, the median age was 45 years (range: 23 to 74); 63% had HCV genotype 1, 7% had HCV genotype 2, 17% had HCV genotype 3, 11% had HCV genotype 4, 2% had HCV genotype 6; 11% had cirrhosis; 84% were male; and 16% were Black.
In EXPEDITION-2, the SVR12 rate in HCV/HIV-1 co-infected subjects was 98% (150/153). One subject experienced on-treatment virologic failure and no subjects relapsed.
14.8 Treatment-Naïve or PRS Treatment-Experienced Adults with Liver or Kidney Transplant without Cirrhosis
MAGELLAN-2 was a single-arm, open-label study in 100 post-liver or -kidney transplant HCV genotype 1, 2, 3, 4, or 6 infected subjects without cirrhosis who received MAVYRET for 12 weeks. The study included subjects who were HCV treatment-naïve or treatment-experienced to combinations of (peg)interferon, ribavirin, and/or sofosbuvir, with the exception of genotype 3-infected subjects who were all treatment-naïve.
Of the 100 subjects treated, the median age was 60 years (range: 39 to 78); 57% had HCV genotype 1, 13% had HCV genotype 2, 24% had HCV genotype 3, 4% had HCV genotype 4, 2% had HCV genotype 6; 75% were male; 8% were Black; 80% of subjects were post-liver transplant and 20% were post-kidney transplant. Immunosuppressants allowed for co-administration were cyclosporine ≤100 mg, tacrolimus, sirolimus, everolimus, azathioprine, mycophenolic acid, prednisone, and prednisolone.
The overall SVR12 rate in post-transplant subjects was 98% (98/100). There was one relapse and no on-treatment virologic failures.
14.9 People Who Inject Drugs (PWID) and those on Medication-Assisted Treatment (MAT) for Opioid Use Disorder
Among 4,655 chronic HCV genotype 1-6-infected adolescents and adults in Phase 2 and 3 trials who received MAVYRET and specified whether or not they had a history of injection drug use, 1,373 subjects were identified as PWID based on self-reported history of injection drug use at trial enrollment and 3,282 subjects did not report injection drug use (non-PWID). Of the PWID population, 62 subjects were considered current/recent PWID (defined as self-reported injection drug use within the last 12 months prior to starting MAVYRET), 959 subjects were considered former PWID (defined as self-reported injection drug use more than 12 months prior to starting MAVYRET), and 352 subjects did not specify current/recent PWID versus former PWID and were not included in the analysis. Compared to former/non-PWID subjects (n=4,241), the current/recent PWID subjects were more frequently male (79%), White (73%), younger (median age [range]: 40 years [19 to 64]), treatment-naïve (94%), and had higher proportions of HCV genotype 3 infection (44%) and HIV co-infection (24%). Similar to the former/non-PWID subjects, the majority of current/recent PWID subjects were non-cirrhotic (73%). The overall SVR12 rate was 98% in former/non-PWID subjects and 89% in current/recent PWID subjects; the difference between the two groups was primarily due to missing data at the time of the SVR12 measurement window in the current/recent PWID group. Virologic failure rates, however, were similar in both groups: 2% in the current/recent PWID subjects and 1% in former/non-PWID subjects.
Among 4,655 chronic HCV genotype 1-6-infected adolescents and adults in Phase 2 and 3 trials who received MAVYRET and specified whether or not they had a history of injection drug use, 225 subjects reported concomitant use of MAT for opioid use disorder and 4,098 subjects reported no use of MAT (332 subjects were not included in the analysis due to missing assessment of MAT). Compared to those not on MAT, subjects on MAT were more frequently male (70%), White (92%), younger (median age [range]: 47 years [23 to 76]), treatment-naïve (89%), and had a higher proportion of HCV genotype 3 infection (50%). Of subjects on MAT, 74% were non-cirrhotic, and 7% were co-infected with HIV, similar to those not on MAT. The SVR12 rates were similar between subjects on MAT (96%) and those not on MAT (98%), with low rates of virologic failure in both groups (<1% and 1%, respectively).
14.10 Clinical Trial in Pediatric Subjects 3 Years and Older
The efficacy of MAVYRET was evaluated in an open-label study (DORA [Part 1 and Part 2]) that evaluated pediatric subjects 3 years to less than 18 years without cirrhosis who received MAVYRET for 8, 12, or 16 weeks. Treatment duration was chosen to match approved adult durations based on HCV genotype and prior treatment experience.
DORA Part 1
Forty-seven subjects were enrolled in DORA (Part 1) and received the adult dose of MAVYRET tablets. The median age was 14 years (range: 12 years to 17 years); the mean weight was 59 kg (range: 32 kg to 109 kg); 55% were female; 74% were White; 13% were Asian, and 9% were Black; 79% had HCV genotype 1, 6% had HCV genotype 2, 9% had HCV genotype 3, and 6% had HCV genotype 4; 77% were HCV TN; 23% were treatment-experienced to interferon; 4% had HIV-coinfection; none had cirrhosis. The overall SVR12 rate was 100% (47/47).
DORA Part 2
Eighty subjects aged 3 years to less than 12 years were enrolled in DORA (Part 2) and received weight-based dosing of MAVYRET oral pellets for 8, 12, or 16 weeks. The median age was 7 years (range: 3 years to 11 years); the mean weight was 26 kg (range: 13 kg to 44 kg); 55% were female; 69% were White, 18% were Asian, and 4% were Black; 73% had HCV genotype 1, 3% had HCV genotype 2, 23% had HCV genotype 3, and 3% had HCV genotype 4; 97.5% were HCV TN; 2.5% were treatment-experienced to interferon; 1% had HIV-coinfection; none had cirrhosis.
Sixty-two subjects received the weight-based recommended dosage. Eighteen subjects received doses lower than the recommended weight-based dosage and were not included in the efficacy assessment. The overall SVR12 rate for the subjects who received the recommended dosage was 98.4% (61/62); the subject who did not achieve SVR12 discontinued treatment due to an adverse reaction [see Adverse Reactions ( 6.1 ) ].
16. How Supplied/Storage and Handling
MAVYRET Tablets
MAVYRET tablets are dispensed in 4-week (monthly) cartons, 8-week cartons or bottles. Each weekly carton contains seven daily dose wallets. Each monthly carton contains four weekly cartons. Each 8-week carton contains 2 monthly cartons. Each child resistant daily dose wallet contains three 100 mg/40 mg glecaprevir/pibrentasvir tablets. Each bottle contains eighty-four 100 mg/40 mg glecaprevir/pibrentasvir tablets. MAVYRET tablets are pink-colored, film-coated, oblong biconvex shaped, debossed with “NXT” on one side.
The NDC numbers are:
- 4-Week Carton: 0074-2625-28
- 8-Week Carton: 0074-2625-56
- Bottle: 0074-2625-84
Store at or below 30°C (86°F).
MAVYRET Oral Pellets
MAVYRET oral pellets are dispensed in child-resistant unit-dose packets in cartons. Each carton contains 28 packets. Each packet contains 50 mg glecaprevir/20 mg pibrentasvir pink and yellow oral pellets. The NDC number is 0074-2600-28.
Store at or below 30°C (86°F).
17. Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Risk of Hepatitis B Virus Reactivation in Patients Coinfected with HCV and HBV
Inform patients that HBV reactivation can occur in patients coinfected with HBV during or after treatment of HCV infection. Advise patients to tell their healthcare provider if they have a history of hepatitis B virus infection [see Warnings and Precautions ( 5.1 ) ].
Risk of Hepatic Decompensation/Failure in Patients with Evidence of Advanced Liver Disease
Advise patients to seek medical evaluation immediately for symptoms of worsening liver problems such as nausea, tiredness, yellowing of the skin or white part of the eyes, bleeding or bruising more easily than normal, confusion, loss of appetite, diarrhea, dark or brown urine, dark or bloody stool, swelling of the stomach area (abdomen) or pain in the upper right side of the stomach area, sleepiness, or vomiting of blood [see Warnings and Precautions ( 5.2 )].
Drug Interactions
Inform patients that MAVYRET may interact with some drugs; therefore, patients should be advised to report to their healthcare provider the use of any prescription, non-prescription medication or herbal products [see Contraindications ( 4 ) , Warnings and Precautions ( 5.3 ) and Drug Interactions ( 7 ) ].
Administration
For MAVYRET oral pellets, advise patients or caregivers to read and follow the Instructions for Use for preparing the correct dose [see Dosage and Administration ( 2.4 , 2.5 )].
Inform patients it is important to take all three tablets at the same time once daily with food as directed. Inform patients that it is important not to miss or skip doses and to take MAVYRET for the duration that is recommended by the physician [see Dosage and Administration ( 2.2 ) ].
If a dose is missed and it is:
- Less than 18 hours from the usual time that MAVYRET should have been taken – advise the patient to take the dose as soon as possible and then to take the next dose at the usual time.
- More than 18 hours from the usual time that MAVYRET should have been taken – advise the patient not to take the missed dose and to take the next dose at the usual time.
Manufactured by AbbVie Inc., North Chicago, IL 60064
MAVYRET is a trademark of AbbVie Inc.
© 2023 AbbVie Inc. All rights reserved.
20082477 R1
Patient Package Insert
| Patient Information | |||
| MAVYRET® (MAV-ih-reht) (glecaprevir and pibrentasvir) tablets | MAVYRET® (MAV-ih-reht) (glecaprevir and pibrentasvir) oral pellets | ||
| What is the most important information I should know about MAVYRET? MAVYRET can cause serious side effects, including: Hepatitis B virus reactivation. Before starting treatment with MAVYRET, your healthcare provider will do blood tests to check for hepatitis B virus infection. If you have ever had hepatitis B virus infection, the hepatitis B virus could become active again during or after treatment for hepatitis C virus with MAVYRET. Hepatitis B virus that becomes active again (called reactivation) may cause serious liver problems including liver failure and death. Your healthcare provider will monitor you if you are at risk for hepatitis B virus reactivation during treatment and after you stop taking MAVYRET. For more information about side effects, see the section “What are the possible side effects of MAVYRET?” | |||
| What is MAVYRET? MAVYRET is a prescription medicine used to treat adults and children 3 years of age and older with:
It is not known if MAVYRET is safe and effective in children under 3 years of age. | |||
Do not take MAVYRET if you:
| |||
Before taking MAVYRET, tell your healthcare provider about all of your medical conditions, including if you:
MAVYRET and other medicines may affect each other. This can cause you to have too much or not enough MAVYRET or other medicines in your body. This may affect the way MAVYRET or your other medicines work or may cause side effects. Keep a list of your medicines to show your healthcare provider and pharmacist.
| |||
How should I take MAVYRET?
| |||
| How should I give MAVYRET oral pellets to my child? See the detailed Instructions for Use for information about how to give or take a dose of MAVYRET oral pellets.
| |||
| What are the possible side effects of MAVYRET? MAVYRET can cause serious side effects, including:
| |||
|
| ||
| The most common side effects of MAVYRET include headache and tiredness. These are not all the possible side effects of MAVYRET. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. | |||
How should I store MAVYRET?
| |||
| General information about the safe and effective use of MAVYRET Medicines are sometimes prescribed for purposes other than those listed in a Patient Information leaflet. Do not use MAVYRET for a condition for which it was not prescribed. Do not give MAVYRET to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about MAVYRET that is written for health professionals. | |||
| What are the ingredients in MAVYRET? MAVYRET tablets Active ingredients: glecaprevir and pibrentasvir Inactive ingredients: colloidal silicon dioxide, copovidone (type K 28), croscarmellose sodium, hypromellose 2910, iron oxide red, lactose monohydrate, polyethylene glycol 3350, propylene glycol monocaprylate (type II), sodium stearyl fumarate, titanium dioxide, and vitamin E (tocopherol) polyethylene glycol succinate. The tablets do not contain gluten. MAVYRET oral pellets: Active ingredients: glecaprevir and pibrentasvir Inactive ingredients: colloidal silicon dioxide, copovidone (type K 28), croscarmellose sodium, hypromellose 2910, iron oxide red, iron oxide yellow, lactose monohydrate, polyethylene glycol/macrogol 3350, propylene glycol monocaprylate (type II), sodium stearyl fumarate, titanium dioxide, vitamin E (tocopherol) polyethylene glycol succinate. The oral pellets do not contain gluten. Manufactured by AbbVie Inc., North Chicago, IL 60064. MAVYRET is a trademark of AbbVie Inc. For more information go to www.MAVYRET.com or call 1-800-633-9110. | |||
| This Patient Information has been approved by the U.S. Food and Drug Administration. 20082477 R1 | Revised: September 2021 | ||
Instructions For Use Section


Package Label. Principal Display Panel
NDC 0074-2625-28
Rx only
Mavyret®
(glecaprevir and pibrentasvir)
100mg / 40mg
Each tablet contains glecaprevir and pibrentasvir 100mg / 40mg
This carton contains 21 tablets packaged as follows:
7 wallets for 1 week of treatment.
Each wallet contains 3 tablets.
Do not use if seal on top of carton is broken or missing
Keep out of reach of children
Store at or below 30°C (86°F)
See Package Insert for full Prescribing Information
AbbVie Inc.
North Chicago, IL 60064
©2020 AbbVie Inc.
abbvie

Package Label. Principal Display Panel
NDC 0074-2625-01
Rx only
Mavyret®
(glecaprevir and pibrentasvir)
100mg / 40mg
Each tablet contains glecaprevir and pibrentasvir 100mg / 40mg
Please see accompanying full Prescribing Information
Each box contains: 1 wallet of 3 tablets each
Keep out of reach of children
Store at or below 30°C (86°F)
See Package Insert for full Prescribing Information
AbbVie Inc.
North Chicago, IL 60064
Product of Ireland
©2020 AbbVie Inc.
abbvie

Package Label. Principal Display Panel

Package Label. Principal Display Panel
NDC 0074-2625-80
Rx only
MAVYRET®
glecaprevir/pibrentasvir tablets
100 mg/40 mg
For Institutional Use Only84 Tablets
abbvie
Do not accept if seal over bottle opening is broken or missing.
Each tablet contains glecaprevir and pibrentasvir 100 mg/40 mg.
Store at or below 30°C (86°F).
See package insert for full Prescribing Information.
AbbVie Inc.
North Chicago, IL 60064 USA
See opposite side of label for Customs Country of Origin.
©2020 AbbVie Inc.
Package Label. Principal Display Panel
NDC 0074-2600-28
Rx only
MAVYRET®
(glecaprevir/pibrentasivir) Oral Pellets
50 mg/20 mg
Each packet contains pink and yellow oral pellets.
Keep out of reach of children.
Store at or below 30°C (86°F).
Recommended Dosage: See Prescribing Information.
AbbVie Inc.
North Chicago, IL 60044
See side of carton for Customs Country of Origin.
©2021 AbbVie Inc.
abbvie
Package Label. Principal Display Panel
NDC 0074-2625-84
Rx only
MAVYRET®
glecaprevir/pibrentasvir tablets
100 mg/40 mg
84 Tablets
Do not accept if seal over bottle opening is broken or missing.
Each tablet contains glecaprevir and pibrentasvir 100 mg / 40 mg.
Store at or below 30°C (86°F).
See package insert for full dosing and Prescribing Information.
See bottom of carton for Customs Country of Origin.
AbbVie Inc.
North Chicago, IL 60064 USA
©2020 AbbVie Inc.
abbvie
